Generation of Optimized Consensus Sequences for Hepatitis C virus (HCV) Envelope 2 Gly-coprotein (E2) by a Modified Algorithm: Implication for a Pan-genomic HCV Vaccine
-
Mohabati, Reyhaneh
-
Department of Molecular Virology, Pasteur Institute of Iran, Tehran, Iran
-
Rezaei, Reza
-
School of Biology, College of Science, University of Tehran, Tehran, Iran
-
Mohajel, Nasir
-
Department of Molecular Virology, Pasteur Institute of Iran, Tehran, Iran
-
Ranjbar, Mohammad Mehdi
-
Department of FMD Vaccine Production, Razi Vaccine and Serum Research Institute, Agricultural Research, Education and Extension Organization (AREEO), Tehran, Iran
-
Samimi-Rad, Katayoun
-
Department of Virology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran
-
 Azadmanesh, Kayhan
Department of Molecular Virology, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 66 953311-20; Email: k.azadmanesh3@gmail.com, azadmanesh@pasteur.ac.ir
Azadmanesh, Kayhan
Department of Molecular Virology, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 66 953311-20; Email: k.azadmanesh3@gmail.com, azadmanesh@pasteur.ac.ir
-
Roohvand, Farzin
Department of Molecular Virology, Pasteur Institute of Iran, Tehran, Iran Tel: +98 66 953311-20 E-mail: farzin.roohvand3@gmail.com, rfarzin@pasteur.ac.ir, k.azadmanesh3@gmail.com, azadmanesh@pasteur.ac.ir, Tel: +98 66 953311-20; E-mail: farzin.roohvand3@gmail.com, rfarzin@pasteur.ac.ir
Abstract: Background: Despite the success of "direct-acting antivirals" in treating Hepatitis C Virus (HCV) infection, invention of a preventive HCV vaccine is crucial for global elimination of the virus. Recent data indicated the importance of the induction of Pan-genomic neutralizing Antibodies (PnAbs) against heterogenic HCV Envelope 2(E2), the cellular receptor binding antigen, by any HCV vaccine candidate. To overcome HCVE2 heterogeneity, "generation of consensus HCVE2 sequences" is proposed. However, Consensus Sequence (CS) generating algorithms such as "Threshold" and "Majority" have certain limitations including "Threshold-rigidity" which leads to induction of undefined residues and insensitivity of the "Majority" towards the "evolutionary cost of residual substitutions".
Methods: Herein, first a modification to the "Majority" algorithm was introduced by incorporating BLOSUM matrices. Secondly, the HCVE2 sequences generated by the "Fitness" algorithm (using 1698 sequences from genotypes 1, 2, and 3) was compared with those generated by the "Majority" and "Threshold" algorithms using several in silico tools.
Results: Results indicated that only "Fitness" provided completely defined, gapless HCVE2s for all genotypes/subtypes, while considered the evolutionary cost of amino acid replacements (main "Majority/Threshold" limitations) by substitution of several residues within the generated consensuses. Moreover, "Fitness-generated HCVE2 CSs" were superior for antigenic/immunogenic characteristics as an antigen, while their positions within the phylogenetic trees were still preserved.
Conclusion: "Fitness" algorithm is capable of generating superior/optimum HCVE2 CSs for inclusion in a pan-genomic HCV vaccine and can be similarly used in CS generation for other highly variable antigens from other heterogenic pathogens.
Keywords:
Amino acids
,
Antibodies
,
Antiviral Agents
,
Consensus sequence
,
Genomics
,
Genotype
,
Hepacivirus
,
Hepatitic C
,
Inventions
,
Phylogeny
,
Vaccines
,
Virus diseases
Introduction :
Hepatitis C virus (HCV) is the primary cause of liver cirrhosis and cancer in humans, affecting an estimated 57 million individuals worldwide (estimated in 2020) 1. Despite the absence of an approved vaccine for preventing HCV infection or its persistence, the introduction of Direct-acting Antiviral Agents (DAAs) in 2012 marked a significant advancement in curing HCV infection that led World Health Organization (WHO) to set a goal to eliminate HCV by 2030 2,3. However, projections indicate that achieving global elimination solely through the use of DAAs without an effective vaccine may not be feasible 3-5. In fact, the number of new HCV infections has been on the rise, more than doubling in the past decade and nearly doubling in the US alone over the last 5 years 6,7. Therefore, development of a vaccine may be crucial in reaching WHO's target of eliminating HCV infection. 5.
The single-stranded RNA (+) genome of HCV encodes for three structural proteins [core, envelope glycoprotein 1 (E1) and E2 and several Nonstructural (NS) proteins]. HCV genome has a high mutation/adaptation rate and displays high genetic heterogeneity which resulted to seven major genotypes and 67 subtypes that differ at the nucleotide levels by 25 to 30% and 15 to 20%, respectively 8. Infection with HCV induces strong humoral and cellular responses against HCV proteins 9. Indeed, induction of efficient neutralizing Antibodies (nAbs) against HCVE2 is shown by both the natural infection and immunization 10. Nonetheless, the high sequence divergence among genotypes and mutation induced-HCV evasion from humoral and cellular immune responses are the major obstacles for development of an efficient vaccine against HCV infection. In fact, HCVE2 contains the most mutating/adapting segments, so called "HyperVariable Regions (HVRs)", in which only 37% of the positions share conserved amino acids across all HCV genotypes, while they (E2-HVRs) harbor the major epitopes needed for induction of nAbs 11. Hence, the challenge lies in inducing cross-genotype (Pan-genotypic) nAbs against HCVE2. Notably, a recent study highlighting the failure of the initial efficacy trial for a prophylactic T-cell viral vector-based HCV vaccine emphasized the crucial requirement for the induction of "broadly reactive Pan-genomic neutralizing Antibodies (bPnAbs)" against HCVE2 12. Therefore, a strategy capable of generating a centralized HCVE2 Antigen (Ag) to induce bPnAbs may greatly aid in the development of a vaccine against HCV infection.13.
The calculation of Consensus (average) Sequences (CS) by determining the most frequently positioned residues/nucleotides within the polypeptides/polymer nucleic acids is an important centralized approach in bioinformatics 14. The generated CS finds numerous applications such as: identification of the functionally related structural motifs in DNA and protein sequences 15,16, design of family-specific degenerate primers 17 and construction of centralized Antigens (Ags) for vaccine formulations targeting highly diverse pathogens like Human Immunodeficiency Virus (HIV) 18,19, influenza 20,21 and Hepatitis C Virus (HCV) 22. To address these concerns, the generated CS should minimize the genetic distances among variable regions of the Ag across strains while preserving the same epitope dominance.
Currently, several bioinformatics tools are available that generate a "CS" from "a set of variable input sequences". These tools use either "Majority" or "Threshold" algorithms 23,24. The "Threshold" selects residues with higher frequency than the user’s selected threshold; whereas "Majority" selects the most common residue in each position (regardless of any indicated threshold). Despite the wide application of these two algorithms, the generation of an intermediate canonical sequence to preserve the features of the original variable regions is still a challenging issue. Indeed, both of these algorithms show several shortcomings that limit their performance for the selection of the desired CSs. In this context, several limitations might be counted for "the threshold-based selection" including: i) Limitation of the 60% frequency for most of the residues, ii) Neglecting residues with lower frequencies than the selected threshold due to the rigidity of this algorithm for the specific threshold point, iii) Induction of the gap positions that result in the information loss within the generated CS. Although the "Majority" algorithm does not have the limitations of the "threshold" but it has another major limitation. It does not consider the evolutionary cost of substituting a specific residue with other candidate residues and only calculates proportions within the sample population. Consequently, choosing between two residues with close frequencies is just based on their frequency differences that may not necessarily reflect the actual frequencies in the population. Therefore, availability of an optimized algorithm to address the aforementioned constraints appears to be essential.
In the present study, first a modified version of "Consensus Generation Algorithm (CGA) " specifically designed for the highly variable HCVE2 Ag was proposed. This algorithm, known as the "Fitness" incorporates residue frequencies weighted by fitness scores derived from BLOSUM matrices (which is the base of the "Threshold and Majority algorithms"). These matrices are commonly used to assess the alignment of protein sequences that have undergone evolutionary divergence 25. Additionally, the HCVE2 CS generated by the "Fitness" algorithm with those generated by the "Majority" and "Threshold" algorithms, were compared considering various parameters such as antigenicity, glycosylation sites, and preservation of epitope dominance.
Materials and Methods :
Calculation of the fitness score: The modified algorithm (Fitness) is based on calculating a fitness score for each residue in a position and selecting the most fitted residue for that position. In this context, the fitness score is calculated by first considering each residue as a possible candidate for that position and subsequent calculation of the tendency of natural selection to keep the residue in that position. This tendency is a function of residue’s frequency and its substitution score, which is obtained from BLOSUMs (Blocks Substitution Matrices). BLOSUM matrix is a substitution matrix used for sequence alignment of proteins based on Local alignment and contains every possible amino acid pair with a quantitative measure of their substitution likelihood 25,26. Moreover, based on the evolutionary distance of the input sequences, the most suitable matrix can be selected (e.g.: Amino acid substitution matrices from protein blocks, 30 for too far sequences and BLOSUM 80 for very close ones). The algorithm multiplies each possible substitution pair score, including the substitution of amino acid with itself and with the frequency of the base amino acid. Subsequently, the total fitness score is calculated by adding each residue’s corresponding scores, and finally, the residue with the highest fitness score will be selected.
The general process for a candidate position can be summarized as follows: i) BLOSUM62 matrix is typically chosen for convenience but BLOSUM 30, 45, 60, and 80 are also viable options, ii) This specific position contains four distinct amino acid residues in different sequences, including ‘G’, ‘W’, ‘T’, and ‘A’, iii) The frequency of residues are ‘G’: 0.142, ‘W’: 0.142, ‘T’: 0.428, and ‘A’:0.285, iv) A table is then created using the letter names as both row and column identifiers. This resulted in each cell of the table representing a potential combination pair, including self-pairs (Figure 1A), v) Next, the substitution score of each pair is retrieved from the BLOSUM62 matrix and placed in its corresponding cells (one cell for self-pairs in the diagonal and two cells for other pairs (Figure 1B), vi) The frequency of each row’s residue is then multiplied by the corresponding substitution scores in that row. For example, the 0.14 frequency of G residue is multiplied in each cell in that row (Figure 1C), vii) Subsequently, the total fitness score is calculated for each residue in each column by adding the corresponding cells of that column (Figure 1D), viii) Finally, the residue with the highest fitness score will be selected as the representative of that position in the final CS.
The above procedure was a detailed explanation for implementing this algorithm in a programming language. However, the fitness score for each residue can be calculated by the following formula where F, R, f, and r denote fitness score function, target residue, frequency function, and each residue including the target itself, respectively.
FR=i=1n(f(ri)(s(R→ri)))
Equation 1 Calculation of the fitness score for each residue. F is: fitness score function, R: target residue, f: frequency function, r: each residue including the target itself, R -> r: substitution of the ‘R’, or target, residue with the ‘r’ residue.
Consensus generation pipeline: The fitness algorithm relies on the creation of a Multiple Sequence Alignment (MSA) as its input. This MSA file should be in a familiar format to let the algorithm extract necessary information such as sequence length, frequency of each residue in any given position, and the list of existing residues in each position. Therefore, to make a consistent pipeline, the alignment part should be added to the base algorithm to make it more robust and reliable.
The MAFFT alignment tool in the python package 27 is chosen as the basic alignment tool for its versatility and numerous adjustment options that play a crucial role in the Fitness algorithm. An essential feature of the MAFFT algorithm is the ability to modify the substitution matrix, which is used in aligning the given sequence set. The substitution matrices in the MAFFT algorithm, which can be selected based on the evolutionary distance among the input sequences, include BLOSUM 30, BLOSUM 45, BLOSUM 62, BLOSUM 80, and other scoring matrices. The selected matrix can be the same or different from the one that is being used in the fitness calculation process (to add extra flexibility to the final pipeline).
The consensus generation pipeline is executed using the Python programming language, incorporating preprocessing steps, score calculation steps, and CS export. All steps in the pipeline are outlined in the flowchart shown in figure 2. The python implementation of the algorithm "bloConGen.ipynb" is provided in the supplementary.
Generation of CSs for HCVE2: In this study, a total of 1698 protein sequences were collected from Genotype 1 (comprising 518 and 438 sequences from 1a and 1b subtypes, respectively), Genotype 2 (comprising 122 and 111 sequences from 2a and 2b subtypes, respectively) and 509 sequences from Genotype 3 (3a) of HCVE2 were retrieved from the "Virus Pathogen Database and Analysis Resource" (ViPR) 28. The high global prevalence of these three genotypes was the main reason for their selection. The utilized sequence databases are provided as supplementary data of the manuscript (HCVE2Db.fasta). The retrieved sequence clusters were aligned using the MAFFT algorithm 29 and employed for the consensus generation process using the "Fitness" in comparison with the other two consensus generating algorithms (Threshold and Majority). Thus, each of the “Fitness and Majority algorithms” were used to generate seven CSs (i.e.: totally fourteen sequences) including five for subtypes (1a, 1b, 2a, 2b and 3a) and two for inter-subtypes of (1a-1b) and (2a-2b). Additionally, "threshold algorithms of: 50%, 70%, and 90%" were employed to create three distinct consensuses for each of the above mentioned seven groups (i.e.: totally twenty one sequences based on threshold algorithms). Therefore, finally thirty five CSs based on all three algorithms (Fitness, Majority, and Threshold) were generated and compared (Supplementary Figure 1; Figure S1). The CLC genomic workbench 5.5 (QIAGEN CLC Main Workbench 5.5, (QIAGEN, Aarhus, Denmark), and BioEdit 30 were used to generate CSs.
Antigenicity prediction and evaluation of the hotspot residues conservation for interaction with nabs: The antigenicity of the generated CSs from the Fitness algorithm, as well as the threshold and majority algorithms, was assessed by comparing them using the "AntigenPro server." Each CS from each algorithm was scored and the Fitness based consensus score was subtracted from it 31. In order to determine the conservation of the Hotspot residues necessary for Ag-nAb interactions in the generated CSs of HCVE2, three well-defined monoclonal anti-envelope bnAbs for HCV neutralization, known as (AP33) 32,33, (1:7) and HC-84.1 34 were considered. The conservation of the Hotspot residues within the interacting epitopes was evaluated by comparing the corresponding amino acids of the reference HCVE2 prototype sequences, primarily identified for interaction of these bnAbs 32-34 and those of the generated CSs of HCVE2 (Supplementary figure 1, Figure S1).
Evaluation of the N-glycosylation sites conservation: The NetNGlyc Server 1.0 35 was utilized to predict the preservation of N-glycosylation sites in the Fitness and Majority based generated CSs. The server's threshold of 0.5 was applied to select sites with greater potency for glycosylation. Subsequently, the number of these sites in the Majority based CSs was subtracted from the Fitness based CSs.
Phylogenetic tree analysis: To assess the status of the "Fitness-generated" CSs compared with that of the "Majority-generated", an unrooted Maximum Likelihood-based phylogenetic Tree was generated for each of the 5 intra subtypes (1a, 1b, 2a, 2b and 3a) and two inter subtypes (1a-1b and 2a-2b) CSs along with reference sequences using MEGA11software 36. The bootstrap method with 1000 replicates was employed for calculation. In addition, to confirm the position of the CSs in the trees, HCVE2 sequences from a database containing 956 HCV E2 sequences for genotype 1 (a), 233 sequences for genotype 2 (b), 509 sequences for genotype 3 (c) along with reference genotypes and subtypes sequences were considered for each tree.
The used HCV reference sequences for various genotypes included: (HCV-G1-NP_671491.1, HCV-G2-YP_001469630.1, HCV-G3-YP_001469631.1) verified in NCBI RefSeq database and confirmed HCV subtype sequences (HCV-1a-AAA45676.1, HCV-1a-AAA4553 4.1, HCV-1b-BAA14233.1, HCV-1b-AAA72945.1, HCV-2a-BAB32872.1, HCV-2b-BAB08107.1, HCV-2a-BAA00792.1, HCV-2b-BAA01761.1, HCV-3a-BA A06044.1, HCV-3a-BAA04609.1) verified by International Committee on Taxonomy of Viruses (ICTV) 37.
Results :
The “Fitness algorithm” generated complete and discrete CSs with evolutionary cost-matched substituted residues: Fitness, Majority, and Threshold (with 50%, 70%, and 90% cut-off) algorithms were employed to generate HCVE2 CSs for intra-subtypes of 1a, 1b, 2a, 2b and 3a and inter-subtypes (1a-1b) and (2a-2b) (Supplementary figure 1, Figure S1). As shown in table 1, "Fitness" algorithm generated complete CSs for all genotypes/subtypes (i.e.: absence of unidentified/undetermined residues). However, several undetermined/unidentified amino acids were positioned in the "Threshold-generated CSs" (lowest and highest for the 50 and 90% thresholds, respectively) that made them inapplicable for further analyses. Therefore, further in silico studies (for antigenic/immunogenic/ glycosylation characterizations) were considered only for HCVE2 CSs generated by "Fitness and Majority" algorithms. But the "Majority" algorithm also generated almost complete CSs for all genotypes with the exception of 1a and 2a subtypes with two and one undetermined/unidentified residues, respectively (Table 1). However, it should be noted that despite generation of almost complete CSs by the "Majority" algorithm (i.e.: absence of unidentified/undetermined residues), there are several residual substitutions for the outputs of this algorithm compared to that of the “Fitness” (Table 2 "substituted Residues" column and Figure S1). The residual substitutions between generated CSs of the two algorithms (Fitness and majority) is more profound in the case of inter-subtype CS "1a-1b" and less for that of the subtypes 1a and 3a. As shown in table 2, there are totally 44 residual substitutions by the "fitness algorithm" (in place of those selected by the "Majority algorithm" for the seven generated CSs. The substitutions correspond to 3, 7, 7, 4, 3, 13 and 7 residues for consensuses generated against 1a, 1b, 2a, 2b, 3a, 1a-1b and 2a-2b subtypes, respectively. Of note, substitutions appear in case of eleven residues with various frequencies including: S, L and A in 12, 9 and 7 substitutions respectively and Q, R and F, V pairs with 3 and 2 substitutions respectively while Y, G, N, T and P residues appear in just one substitution (Table 2, Figure S1).
The "Fitness"-generated HCVE2 CSs showed high antigenicity scores and preserved the critical residues for nAb interactions: The HCVE2 CSs generated by the "Fitness" and "Majority" algorithms were assessed for their antigenicity potential using the AntigenPro server 31. Comparing the "Fitness and Majority" algorithms indicated that antigenicity scores above the server threshold level (0.5) were obtained for CSs generated by both algorithms (Table S1) but still higher values were observed for that of the "fitness" (Table 2 "Antigenicity column" and Table S1). Moreover, present results indicated that similar to the "Majority", critical/hotspot residues in both linear and conformational epitopes that are needed for induction/interaction of well-known nAbs: AP33 (L413, N415, G418, W420), HC84.1 (L441, F442) and 1:7 (G523, T526, Y527, W529, G530, D535) (32-34) are preserved (i.e.: not substituted) in the HCVE2 CSs generated by "Fitness" algorithm (Figure S1, Table 2 "column of Substituted Residues").
The "Fitness"-generated CSs preserved all glycosylation sites within the HCVE2: The "Fitness" generated consensus HCVE2 and that of the "Majority" in the NetNGlyc server (35) for the presence of glycosylation sites were evaluated. As inferred from table 2, table S1, while "Fitness and Majority algorithms" generated CSs with almost the same number of the preserved glycosylation sites, "fitness" still showed superior performance in case of 1b subtype of HCVE2 (Table 2, Table S1).
The position of the "Fitness"-generated CSs was preserved in the phylogenetic tree: Figure 3 displays unrooted Maximum Likelihood-based phylogenetic trees for five HCV subtypes (1a, 1b, 2a, 2b & 3a) and two inter subtypes (1a-1b & 2a-2b) were rendered through MEGA11software 36 (generally seven representative trees). The Sequence Identity Matrix (SIM) calculated by BioEdit software 30, indicated that the distance of Majority and Fitness-generated CSs were closely similar and comparable to the HCVE2 sequence database (Table S2). The results of the phylogenetic analyses for HCVE2 sequence database including 956 HCV E2 sequences for G1 (a), 233 sequences for G2 (b), 509 sequences for G3 (c) confirmed the SIM results. These analyses utilized HCV reference sequences (obtained from NCBI RefSeq database and confirmed subtypes obtained from ICTV 37 along with the fitness/majority generated CSs (Figure 3, Figure S2) also confirmed the SIM results indicating that the overall position of all CSs produced by either "Fitness" or "Majority" is preserved on the tree.
Discussion :
In the present report, the objective was to generate superior CSs of HCVE2 antigens as potential vaccine candidates. Initially, the "Fitness" algorithm, which is a modification of the "Majority algorithm" based on BLOSUM matrices, was introduced. This algorithm focuses on fitness scores. Subsequently, the CSs of HCVE2 generated by the fitness algorithm were compared to those generated by the threshold (50,70,90) and majority algorithms for various parameters including the frequency of undetermined/unidentified residues, antigenicity, preservation of epitope dominance, glycosylation sites, and overall position on the phylogenetic tree. The selection of five intra subtypes (1a, 1b, 2a, 2b, and 3a) and two inter subtypes (1a-1b and 2a-2b) belonging to HCV genotypes one to three was based on the high global prevalence of these genotypes. The findings demonstrated that the CSs generated by the "Fitness" algorithm overcame the limitations of the currently available algorithms for CS generation ("Threshold" and "Majority") by providing a completely defined, gapless sequence (without any occurrence of undefined residues, which is a common issue in the "Threshold" algorithm), while also considering the evolutionary cost of amino acid substitution (which is a limitation of the "Majority" algorithm). Furthermore, the consensus HCVE2 sequences generated by the fitness algorithm exhibited superior antigenicity and preservation of glycosylation sites, and their positions in the phylogenetic trees were also maintained. Since "fitness" algorithm is a modification (improved version) of the "Majority" algorithm, it can be similarly used to create "consensus sequences for antigens from other pathogens as shown for: Influenza H1N1 38, H5N1 39, H3N2 40, HCV E2 (NOTC1 and NOTC2) 41 and HIV-1 42 and similar to the "Majority" the range of BLOSUM 30-80 can be applied based on the variability of the pathogen of interest as a modification.
Based on the data presented in table 1 (sequence comparison column) and figure S1, it is evident that only the "Fitness algorithm " was able to produce well-defined, gapless consensus HCVE2 sequences. In contrast, the "Threshold-generated HCVE2 CSs" contained numerous undefined residues, particularly at higher cut-off values (e.g. T90 had the highest amount of undefined residues, while T50 had the lowest). The "Majority" algorithm also resulted in undetermined or unidentified residues in the consensus sequences of HCV 1a and 2a subtypes. Previous studies have utilized the Majority algorithm for generating CS antigens of HIV Env 18,19,43 and Influenza hemagglutinin 21,38-40,44 proteins. Additionally, the "Threshold algorithm" in UGENE software 23 has been used for generating CS antigens for Dengue virus 45 and SARS-CoV2 46 envelope proteins. However, the present study found that the "Threshold algorithm" was unsuccessful in generating complete CSs for highly divergent E2 proteins across HCV genotypes or subtypes, in contrast to the "Fitness algorithm". The obvious explanation for this result lies in the higher heterogeneity of the HCVE2 in comparison to the HIV Env, Influenza hemagglutinin proteins and antigens of Dengue and SARS-CoV2 viruses. In fact, the HCV HVR1 exhibit only one amino acid with 100% conservancy among all six HCV genotypes 11 as evidenced by the 1698 HCVE2 sequences retrieved from the present database (supplementary file: HCV-Db.fasta). Notably, there is over 80% variability at a single position within the 437 sequences of HCVE2-subtype 1b (calculated data is not shown). Consequently, the significant variability in HCVE2 poses a formidable challenge for the "Majority and Threshold algorithms," particularly when attempting to generate a CS from multiple HCV genotypes. In this context, a recent report has described the use of the "Majority algorithm" to create CSs from genotype 1 (1a-1b) HCVE2 (referred to as NOTC1 and NOTC2 CSs) 41. However, apparently, to be able to use the "Majority" for generation of NOTC1 and NOTC2CSs, the HVR1 regions from highly variable HCVE2 (1a-1b) sequences were removed. This decision was made despite the fact that HCVE2-HVRs harbor the major epitopes necessary for induction of neutralizing antibodies (nAbs) 47,48. Moreover, In spite of the fact that most of the Majority-generated HCVE2 CSs do not have unidentified or undetermined residues, there are totally 44 residual substitutions for the outputs of this algorithm compared to the "Fitness". These substitutions can be observed in table 2 "Substituted Residues" column and figure S1. Out of the seven generated consensuses, the Majority algorithm has selected eleven different residues. Among these, residues S, L, and A have 12, 9, and 7 substitutions respectively. Additionally, the pairs Q, R, and F, V have 3 and 2 substitutions respectively, while Y, G, N, T, and P have only one substitution each. These substitutions occur because the Fitness algorithm takes into account the "Evolutionary Cost" based on the obtained score. Indeed, "Fitness algorithm" weights amino acid frequencies by substitution costs from BLOSUM matrix which in these cases overwrites amino acid selection by Majority algorithm. For example, the Majority algorithm selected residues S and L in 12 and 9 different positions within the generated HCVE2 CSs, but the Fitness algorithm assigned them a lower score for selection, resulting in their replacement with other amino acids (Table 2 "Substituted Residues" column and Figure S1). Hence, the "Fitness algorithm" could potentially overcome the limitations of the "Majority and Threshold algorithms" in generating of the reliable CSs against highly variable Ags likeHCVE2.
As shown in figure S1, results indicated that the critical/hotspot residues in both linear and conformational epitopes were perfectly preserved in the "Fitness-generated HCVE2 CSs" as well as in those generated by the majority algorithm (Table 2 "Antigenicity column"). Presence of these critical/hotspot residues is essential for interaction of prominent nAbs such as: AP33 (L413, N415, G418, W420), HC84.1 (L441, F442) and 1:7 (G523, T526, Y527, W529, G530, D535) 32-34. Preserving the specific epitopes through the conservation of their contributing critical/hotspot residues is an important and indispensable feature of an algorithm for the generation of consensus Ags. Therefore, the "Fitness" might be considered as a reliable algorithm for the generation of a consensus vaccine Ag against highly variable pathogens and HCV types/subtypes. In agreement with our results, prior studies on Influenza Hemagglutinin (H1N1) consensus protein 38 indicated the capability of "Majority" algorithm in the generation of CSs with preserved critical/hotspot residues for variable antigens. Accordingly, the two HCVE2 consensus proteins (NOTC1 and NOTC2), generated by the "Majority algorithm" from 1a and 1b subtypes of genotype 1 41 also retained their binding capabilities to well-known HCV nAbs including: AP33 32,33 and 1:7 34. However, it should be noted that still the Fitness-generated HCVE2 CSs showed higher antigenic scores compared to that of the majority algorithm (Table S1) indicating its superiority in this context.
Besides critical residues that interact with nAbs, natural glycosylation of the HCVE2 glycoprotein might play important structural roles for the protein as an Ag 49. As shown in table 2 (Number of Glycosylation sites column) and table S1, while "Fitness, Majority and T50 algorithms" generated CSs with almost the same number of the preserved glycosylation sites, but still "fitness" showed superior performance for 1b subtype of HCVE2. Consistent with the present study results, a prior study on Influenza H3N2 consensus protein also showed that all glycosylation sites in the "Majority-generated consensus" proteins were preserved in the same manner as natural strains 40. But in contrast, for the HIV-env protein, the generated CSs by both the "Threshold" and Majority algorithms led to an increased number of glycosylation sites compared to that of the natural protein. In vivo immunization studies using the Majority-generated consensus HIV-env proteins indicated that increased number of glycosylation sites resulted in the shielding of the non-neutralizing or poorly conserved epitopes and thus improved the exposure of the conserved, neutralizing epitopes to the immune system 50. However, on the contrary, it is shown that preservation of two amino acids involved in glycosylation of an Influenza H1N1-generated CS resulted to the masking of important epitopes involved in Ag-Ab interactions 38. Taken together, prediction of the immunization effect of the glycosylation site preservation (i.e.: augmentation, or decline of the immunogenicity) in the generated consensus Ags needs further in vivo investigations.
The phylogenetic tree analyses indicated that the distance between the "Majority and Fitness-generated CSs" were closely similar and comparable to that of the HCVE2 sequence database (Table S3) and thus the position of the "Fitness"-generated CSs were preserved (Figure 3, Figure S2). These results suggest that despite the "Fitness" algorithm introducing several residual substitutions compared to the "Majority" algorithm (that resulted to the enhancement of antigenic/immunogenic characteristic and consideration of the evolutionary characteristics of HCVE2 as an Ag), but still its position on the phylogenetic tree is conserved. This observation highlights the valuable contribution of the "Fitness" algorithm in generating CSs against highly variable and heterogeneous proteins. Consistent with the present study results, prior studies on the generated CSs by "Majority" for Influenza H1N1 38, H5N1 39, H3N2 40, HCV E2 (NOTC1 and NOTC2) 41 and HIV-1 42 also indicated the preservation of their positions on the phylogenetic tree.
Recently, by comparing various in silico methods to identify T-cell based CD4+/CD8+ epitopic peptides in HCVE2 of various genotypes, two evolutionary conserved peptides notified as P2 (VYCFTPSPVVVG) and P3 (YRLWHYPCTV) were identified 51. The present study, which focused on generating the complete HCVE2 CSs to induce nAbs, cannot be directly compared to this report. However, it is worth noting that both the P2 and P3 T-cell based CD4+/CD8+ peptides mentioned in the report are also present in our "Fitness-generated CSs". This observation further confirms the capability of the "Fitness algorithm" to preserve the conserved residues within the whole generated HCVE2 CSs that are involved in the induction of the cellular immunity, too.
More recently, another immunoinformatics method was also applied to design a multi-epitope vaccine against HCV. To this end, several B- and T-cell epitopes from conserved regions of the E2 protein of seven HCV genotypes were joined together with cholera enterotoxin subunit B (CtxB). In silico analyses and structural predictions indicated binding stability with Toll-like receptor 2 (TLR2) and TLR4 52. However, it is important to note that this study also focused on selecting and combining conserved epitopes, which differs from our approach of generating the complete HCVE2 protein CSs to induce nAbs. Similarly, in another recent report, various immuno-informatics tools and bioinformatics databases were deployed to identify potential consensus B-cell and T-cell epitopes from spike glycoprotein of "Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-COV-2)", the virus responsible for the latest global pandemic. However, consistent with the prior study, only separate epitopes and not the whole spike glycoprotein were taken into account for consensus generation, rather than the entire spike glycoprotein 53. It should be also noted that Position Specific Scoring Matrix (PSSM) and similar matrixes which are dependent on the residual position (in contrast to, BLOSUM that is position-independent) are mostly used for "BLAST and motif searches and prediction" applications in the form of the "relative consensuses with probable percentages of the occurrence of a residue in a specific position (which is derived from a multiple sequence alignment)" rather than creation of the exact consensus for a whole protein which is going to be expressed and used as an antigen 54,55. Therefore, to our best of knowledge, to date, only BLOSUM-based algorithms (such as threshold and majority) have been used in studies related to consensus generation of antigenic proteins for various pathogens 18,19,21,23,32-34,38-41,43,46.
Finally, it is worth mentioning that when capturing the most common sequences in Ag databases, there is a potential sampling bias in the generated CS towards the antigenic cluster that has been frequently isolated and reported 38,39. To avoid this sampling bias, a layered consensus building approach has been adopted for the generation of HCVE2 protein CS in genotype 1. However, the HVR1 region has been excluded due to limitations in the algorithm. Thus, combination of the layering approach and the Fitness algorithm along with the inclusion of other HCV genotypes can expand the breadth of the CS induced immunity.
Conclusion :
In summary, a modification based on BLOSUM matrices (Fitness-score oriented) to the "Majority algorithm" which overcame the limitations of threshold and majority algorithms in generating CSs from highly variable proteins like HCVE2 was successfully implemented. The Fitness-generated HCVE2 sequences exhibited superior antigenic, immunogenic, and evolutionary characteristics compared to those generated by the Threshold/Majority algorithms, while still maintaining their positions in phylogenetic trees. These promising results from authors in silico study suggest that the consensus HCVE2 Ag sequences generated by the "Fitness" algorithm could serve as potential vaccine candidates for producing cross-protective neutralizing antibodies, warranting further investigation in future in vitro and animal studies. These generated consensuses could be utilized in vaccine development, similar to multivalent vaccines using various platforms. Additionally, the "Fitness" algorithm could be applied to optimize CSs for other highly variable antigens from diverse pathogens.
Supplemented Materials :
The python implementation of the "Fitness" algorithm is provided in the supplementary (bloConGen.ipynb). For the execution of the "jupyter notebook file", the "Biopython Pandas and Mafft Commandline" packages should be installed in the python environment. The database of aligned sequences used to generate Threshold, Majority and Fitness CSs is also provided in the supplementary (HCVE2Db.fasta). The supplementary figures (Figures S1, S2) and tables (Table S1, S2) are also provided as a file in the supplemented materials (Supplementary Figures and Tables).
The unrooted Maximum Likelihood-based phylogenetic Trees for three HCV genotypes including HCVE2 sequence database of 956 HCV E2 sequences from genotype 1 (G1), 233 sequences from genotype 2 (G2) and 509 sequences from genotype 3 (G3) along with confirmed genotypes and subtypes and CSs were provided as Figure S2.
Acknowledgement :
This study was in partial fulfillment of the Ph.D. degree for RM in the graduate school of Pasteur Institute of Iran. Some information related to the modified algorithm was partially presented as a preprint draft in "bioRxiv; the preprint server for biology".
Funding: This study was partially supported by grant No. 1968 from research council of the Pasteur Institute of Iran and education office of this institute (grant no. 1255).
Conflict of Interest :
The authors declare that they have no competing financial interests.
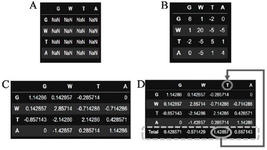
Figure 1. Calculation of "fitness score" for each amino acid position and selection of the most fitted residue. A) The blank (unfilled) table. B) Each cell contains the corresponding substitution score of its row and column pair. C) Each cell in a row has been multiplied by the frequency of its corresponding row name. D) The last column contains the total of each column. The maximum fitness score and its corresponding column names are highlighted.
|
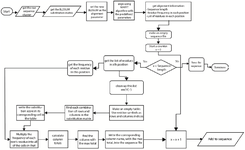
Figure 2. The consensus generation pipeline for the implementation of the consensus generation. The Flowchart illustrates the preprocessing steps, the score calculation steps, and the generated final CS. The python implementation of the Fitness algorithm “bloConGen.ipynb” is provided in the supplementary.
|
![<p>Figure 3. Phylogenetic tree analyses. Three unrooted Maximum Likelihood-based phylogenetic trees for HCV CSs generated by Fitness and Majority algorithms from every five subtypes (1a, 1b, 2a, 2b & 3a) and two inter subtypes (1a-1b & 2a-2b) were rendered through MEGA11software and calculated by the bootstrap method using 1000 replicates [34] using HCV reference sequences for each genotype and confirmed subtypes: a: 1a, b: 1b, c: 2a, d: 2b, e: 3a, f: 1a-1b, g: 2a-2b (■: Majority, ●: Fitness).</p>](Images/Articles/60594/f3_small.png)
Figure 3. Phylogenetic tree analyses. Three unrooted Maximum Likelihood-based phylogenetic trees for HCV CSs generated by Fitness and Majority algorithms from every five subtypes (1a, 1b, 2a, 2b & 3a) and two inter subtypes (1a-1b & 2a-2b) were rendered through MEGA11software and calculated by the bootstrap method using 1000 replicates [34] using HCV reference sequences for each genotype and confirmed subtypes: a: 1a, b: 1b, c: 2a, d: 2b, e: 3a, f: 1a-1b, g: 2a-2b (■: Majority, ●: Fitness).
|
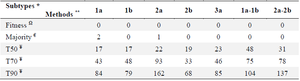
Table 1. Comparison of the number of undetermined residues in the HCVE2 CSs generated by "Fitness, Majority and Threshold algorithms"
* HCVE2 subtypes used to generate CSs
** The digits indicate the number of undetermined/unidentified residues in the generated HCVE2 CSs by each algorithm for the indicated subtypes. Please see "supplementary figure 1 (Figure S1)" for the exact position of the undetermined/unidentified residues within the generated CSs.
Ω "Fitness" algorithm generated complete CSs for all HCVE2 genotypes/subtypes (i.e.: absence of unidentified residues)
€ "Majority" algorithm generated complete CSs for all HCVE2 genotypes with the exception of 1a and 2a subtypes with two and one undetermined/unidentified residues, respectively.
¥ The threshold rigidities are denoted by T50, T70 and T90. Threshold-based generated CSs had more unidentified residues in higher cut-off values (i.e.: highest for T90 and lowest for T50).
|
Table 2. Comparison of the various aspects of the HCVE2 Fitness-based CSs and that of the Majority”
Ω Substituted residues in the generated CSs by "Fitness (preceding residue)" compared to that of the “Majority (following residue)” algorithm in the specified positions (the indicated digit between two residues) denote the consideration of the evolutionary cost by the “Fitness” algorithm (Figure S1). Fitness residual substitutions for eleven different selected residues of majority algorithm including: S, L and A in 12, 9 and 7 substitutions respectively (shown by underlined, underlined/bold and bold, respectively) and Q, R and F, V pairs with 3 and 2 substitutions respectively and Y, G, N, T and P with just one substitution are provided.
*Digits indicate the differences (subtractions) of the antigenic scores for “Fitness” generated CSs and that of the “Majority” showing the higher values for the preceding one (Table S1).
** Digits indicate the differences (subtractions) of the number of the glycosylation sites for the “Fitness” generated CSs and that of the “Majority” showing almost similar values for both (Table S1).
|
|