High Expression of Methylotrophic Yeast-Derived Recombinant Human Erythropoietin in a pH-Controlled Batch System
-
Maleki, Ahmad
-
Faculty of Pharmacy, Tehran University of Medical Science, Tehran, Iran
-
Research and Production Plant, Pasteur Institute of Iran, Karaj, Iran
-
 Roohvand, Farzin
Ph.D., Hepatitis & AIDS Department, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 21 66969291 Fax: +98 21 66969291 E-mail:rfarzin@pasteur.ac.ir farzin.roohvand@gmail.com
Roohvand, Farzin
Ph.D., Hepatitis & AIDS Department, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 21 66969291 Fax: +98 21 66969291 E-mail:rfarzin@pasteur.ac.ir farzin.roohvand@gmail.com
-
Hepatitis & AIDS Department, Pasteur Institute of Iran , Tehran, Iran
-
Khanahmad, Hossein
-
Research and Production plant, Pasteur Institute of Iran , Karaj, Iran
-
Beiruti, Ahmad
-
Research and Production plant, Pasteur Institute of Iran , Karaj, Iran
Abstract: To accomplish the worldwide demand for recombinant human erythropoietin (rHuEpo) as a therapeutic, application of cost-efficient expression system of methylotrophic yeast Pichia pastoris (P. pastoris) rather than mammalian cells is indispensable. Herein, a report on high levels secreted-expression of Pichia-derived rHuEpo by batch fermentation in a pH stabilized format is presented. The full length cDNA of rHuEpo was inserted into pPICZaA vector under control of AOX1 promoter, downstream of the secretion-a-factor and electroporated into P. pastoris strain X33. The highest expression transformant was selected by screening among the colonies surviving high concentration of Zeocin (1.0 mg/ml), followed by comparative small scale expression analysis by ELISA. Stabilization of pH around 6.0 by adding phosphoric acid into the culture media during induction period, improved the yield of expression to 150 mg/l of the media. Single-step Nickel-affinity chromatography was employed for purification of rHuEpo-6xHis to 80% purity. Analyses by SDS- PAGE, Western blot and N-terminal protein sequencing confirmed the authenticity of the 33 kDa expressed rHuEpo with a native N-terminal indicating the proper cleavage of secretion-signal. Results of this study, further confirmed the possibility of employing methylotrophic yeast for scaled up production aims of rHuEpo as a cost-efficient expression system when provided evidence for higher expression yields through application of pH-controlled systems.
Introduction :
Erythropoietin (Epo), a 30-38 kDa glycoprotein hormone (exact Molecular Weight-MW depends on degree of glycosylation) is produced mainly by kidney as a 193 amino acids pre-mature protein which stimulates the growth of red blood cells and as a consequence increases haemoglobin levels (1, 2). Administration of Epo as a therapeutic improves the quality of life in patients with cancer, renal and heart failure anemia (3).
To fulfil the high medical demand for Epo globally, large scale production of recombinant Human Epo (rHuEpo) was originally initiated in Chinese Hamster Ovary (CHO) cells (1). Nonetheless, due to cost-efficiency considerations for scaled up production, more efficient heterologous expression systems, rather than mammalian cells for this valuable drug are being explored.
To this end, production of Epo in different prokaryotic hosts like Escherichia coli (E. toli) (4) and Bacillus brevis (5), as well as eukaryotic hosts such as Saccharomyces cerevisiae (S. cerevisiae) (6) and Drosophila melanogaster (7) was described. However, since proper glycosylation of Epo as a hormone is important for some of its functional properties such as biological half-life (8), neither prokaryote systems lacking the ability to glycosylate proteins nor microbial eukaryotes like S.cerevisiae which hyperglycosylates proteins (9) could be considered as alternatives to currently exploited CHO cells for production of rHuEpo.
In recent years, a few yeast species capable of growing on methanol as sole carbon and energy source have been identified and their application in biotechnology for high level and cost-efficient production of recombinant proteins with proper post-translational modifications were established (10). These so called methylotrophic yeasts belong to four genera; Hansenula, Pichia, Candida and Torulopsis and their most prominent representatives are Hansenula polymorpha (H. polymarpha), Pichia pastoris (P. pastoris), Candida boidinii and Pichia methanolica, respectively (11). While H. polymorpha and P. pastoris are distinguished as industrial platforms for heterologus protein production, the other two methylotrophic yeasts are still in the research level of applications (12).
Currently, the heterologous protein expression system of methylotrophic yeast “P. pastoris” is broadly used for production of many human and therapeutic proteins in biopharmaceuticals industries (13). In fact with the advantages of both prokaryotic and eukaryotic systems and the availability of strong promoter for alcohol oxidase, AOX1, which is tightly regulated and induced by addition of methanol to the growth medium, P. pastoris provides the potential for producing correctly folded and properly glycosylated recombinant proteins. It often allows the recombinant protein to be secreted in large quantities when it does not secrete significant amounts of intrinsic proteins, a phenomenon that facilitates downstream purification steps (14).
However, to transport the expression profile of a new protein from research scale to industrial levels, the possibility of high level expression and purification yields should be proved. In this context, recently, expression of rHuEpo by P. pastoris in a batch culture system using baffled flasks is also reported (2); but the expression level was shown to be as low as 5 mg/l. Therefore, for scaled up- production purposes, there is absolute need to improve expression/culturing protocols for much higher yields of rHuEpo expression in methylotrophic Pichia.
Such optimizations may be achieved by exploring better vectors, host strains, and expression clones as well as optimizing the fermentation process to improve the yield and stability of expressed proteins by controlling the medium composition, pH, temperature, methanol induction and feed mode (15). To this end, more recently higher production levels for P.pastoris-derived rHuEpo (130 mg/l) by Fed-batch methanol feeding strategy in the presence of co-substrate sorbitol
Materials and Methods :
Construction of expression vector for secretion of rHuEpo in methylotrophic yeast
The synthetic cDNA encoding for the 166 native amino acids of the homo sapiens Epo gene (NCBI accession no. NM_000799) in tandem with the DNA sequence of six histidine amino acids (6xHis-tag) located at the C-terminal of Epo which was synthesized and cloned in pUC57 plasmid (Bio Basic Inc, Ontario, Canada) was used as template in PCR reactions throughout this study. For further direct cloning of PCR-amplified products in the Pichia expression vector pPICZaA (Invitrogen), Xho I and Xba I restriction sites were considered in forward (F-EPO: 5' CTCGAGAAAAGAGCCCCACCACGCCTCATC3') and reverse (R-EPO: 5' TCTAGATTATCAATGATGATGATGATGATGTCTGTCCCCTGTCCT 3') primers, respectively (restriction sites are underlined). This cloning strategy provided the Kex2 cleavage site at the 5’ ends of the DNA encoding Epo to locate the amplified fragment in frame with secretion sequence (a-factor) under control of AOX1 promoter while the 6xHis-tag and two termination triplets were located at the 3’ ends in tandem (Figure 1).
DNA amplification was carried out through 30 cycles of denaturation (30 sec at 94°C), annealing (35 sec at 58°C) and extension (60 sec at 72°C), followed by a final elongation (10 min at 72°C) in a Perkin–Elmer thermocycler. Following digestion by Xho I and Xba I restrictions enzymes and gel purification (QIAquick Gel Extraction Kit, QIAgen), PCR-amplified fragments and PICZaA vector were mixed in the ligation reaction. The product of ligation reaction was used to transform E. coli TOP10 competent cells (Invitrogen) and developed Zeocin resistant bacterial colonies were screened for the presence of the proper recombinant construct.
The presence and accuracy of the inserted gene within the expression cassette in the final recombinant PICZaA-Epo construct was confirmed by both restriction analyses using Xho I and Xba I enzymes and DNA sequencing reactions using the 5'AOX1(5’-GACTGGTTCCAATTGACAAGC-3’) and 3'AOX1(5’-GCAAATGGCATTCTGACATCC-3’) primers (Invitrogen) which amplify the expression cassette together with the cloned Epo gene.
Electro-transformation of methylotrophic yeast and selection of expression clones
Around 10 µg of purified PICZaA-Epo plasmids (using QIAprep Spin Miniprep Kit) were linearized with Sac I enzyme (Fermentas) and electroporated into the yeast P. pastoris, wild-type host strain X-33 (Invitrogen) in 0.2 cm cuvettes at 1.5 kV (25 µF and 200 O), using a Bio-Rad Gene Pulser according to supplier’s instructions (Easy Select Pichia Expression manual, Invitrogen).
Following electroporation, cells (200 µl) were plated on YPDS medium (1% Yeast extract, 2% Peptone, 2% Dextrose, 1 M Sorbitol) supplemented with 100 µg/ml Zeocin and incubated for 3-5 days at 30°C. Zeocin-resistant yeast transformants were further transferred on plates containing 1 mg/ml Zeocin. Finally, colonies that survived this concentration of Zeocin were screened for confirming the methanol utilization positive phenotype (Mut+) according to the supplier’s instructions (Invitrogen).
Briefly, a single colony of each Zeocin-resistant transformants was first streaked on a MM (Minimal Methanol) plate and then on a MD (Minimal Dextrose) plate and incubated 2 days at 30 °C. Mut+ transformants were identified through their normal growth on both plates within this time (in these conditions MutS strains only grow on the MD plate and show little or no growth on the MM). From screened Mut+ transformants that survived a concentration of 1 mg/ml Zeocin, 10 colonies were randomly selected and confirmed for the presence of the corresponding Epo gene by PCR using AOX1 primers. Finally, confirmed Mut+ transformants were analyzed for expression levels in a mini scale (50 ml conical tubes containing 10 ml of culture media) and the highest expression clone was selected on the basis of ELISA, Western blot and densitometric assays on the culture supernatants as described in th
Result :
PICZaA-Epo expression vector and selection of yeast transformants
The human cDNA of Epo which was employed in this study encoded 166 native amino acids of the protein. In our cloning strategy, using Xho I and Xba I sites of pPICZaA, the cDNA of Epo was cloned in frame with alcohol oxidase (AOX1) transcription/ translational cassette. This strategy inserts the Epo cDNA downstream of the prepro-a-factor sequence and immediately after the Kex-2 signal cleavage site to express and secrete Epo into the culture media with a native N terminus. Furthermore, two stop codons were placed immediately after the 6xHis-tag codons for preventing of any ribosomal-pass through (Figure 1).
The restriction analyses of the constructed recombinant Epo plasmid (Figure 2) and DNA sequencing analysis demonstrated the correct insert orientation and complete accuracy for the PICZaA-Epo construct without any unwanted mutations.
Due to the advantages of electroporation, such as high frequency of transformation (especially possibility of multi-copy insertion), this method was employed to transform yeast cells by the constructed plasmid (PICZaA-Epo).
To reach the highest possible expression levels for our gene of interest (Epo), we primarily tried to select transformants that potentially may contain multiple copies of the integrated vector. For this purpose, transformants that appeared after 3-5 days on plates containing 100 µg/ml Zeocin were further transferred on plates containing 1 mg/ml Zeocin. Colonies that survive on such a high concentration of Zeocin, potentially may contain multi copies of the cassette inserted into their genome (19). Moreover, the highest expression clone for scale-up experiment was screened from 20 randomly selected Mut+-Zeocin resistant transformants (1 mg/ml) by culturing them in small scale (10 ml) for comparatively appraising the level of expression among them. It should be noted, however that multiple cassette insertions into the genome of selected transformants should be verified by methods like Southern blotting.
Optimization of media and buffering system of fermentation system for high level expression
Following the selection of best expression-clone, scaled-up expression of Epo in unbuffered media (simple: 1.34% YNB, 4 x 10-5% biotin, 0.5% methanol) and (complex: 1% yeast extract, 2% peptone, 1.34% YNB, 4 x 10-5% biotin, 0.5% methanol) was tried, but the expression levels were lower than 1 mg/l. This low level expression may be explained by shocking levels of [H+] concentration during yeast fermentation (20). Shocking levels of [H+] may be attributed to extreme high or low concentrations of H+ that could inhibit the expression of the protein during fermentation.
In fact, by the end of fermentation, utilization of unbuffered simple medium resulted in extreme acidic conditions (pH=3.5), apparently due to production of methanol oxidized metabolites when on the contrary to simple media, the pH for the unbuffered complex medium resulted in extreme basic conditions (pH=8.5) apparently due to the production of ammonium via metabolizing amino acids existing in yeast extract and peptone. This finding for extremely low expression levels of Epo in unbuffered media prompted us to evaluate the effect of different pH values on the level of Epo expression.
In this context, by adjusting the pH value in a range of 5.0 – 7.0 (with 1.0 interval) we found that expression of Epo in buffered methanol complex medium with a pH=6.0 was higher than other pH ranges (Figure 3a). Moreover we could show that stabilizing the pH at 6.0 throughout the expression time by addition of 0.1 M phosphoric acid in a batch format every 24 hrs, led to a considerable increasing of Epo expression level (150 mg/l) compared to unstabilized pH conditions (120 mg/l) (Figure 3b).
It should be noted that a perfect stabilization of pH may be achieved only through application of an automatically-controlled fermenter that adjust the pH through continuous m
Discussion :
Although, expression of rHuEpo by P. pastoris in a batch culture system using baffled flasks was demonstrated (2), their reported expression level was as low as 5 mg/l which is not an appreciated yield for scaled-up production aims.
The present study was undertaken to investigate possibility of application of other fermentation strategies rather than fed-batch methanol feeding and sorbitol utilization for high yield production of rHuEpo by P.pastoris (130 mg/l) which was reported recently (16) and to confirm possibility of scaled up production of rHuEpo by methylotrophic yeast. To our knowledge, this is the second report on high level expression of rHuEpo in methylotrophic yeast when the first report on optimization of fermentation process through stabilizing the pH and from a national point of view it is the first report on home-made production of yeast-derived rHuEpo.
While described capacities for recombinant protein production by P. pastoris varies from 1 to 10000 mg/l (15), expression levels for rHuEpo reported in the present study (150 mg/l) provides sufficient deserve for future large scale plans on methylotrophic yeast-derived Epo production. In fact, by optimizing other factors involved in the fermentation process of methylotrophic yeasts such as temperature, induction mass, methanol concentration and feed mode through application of a controlled-continuous culturing system it may be possible to attain much higher yields for scaled-up applications (15). Moreover, by employing other strategies like addition of detergents such as different Tweens (20, 40, 60, 65 or 80) and/or addition of co-substrates such as sorbitol even higher yields of expression may be achieved (16, 21).
Generally high level expression of a gene of interest depends upon the generation and detection of a recombinant strain that contains multiple copies integrated at the AOX1 locus (22). Because of the low frequency of multiple gene insertion events (between 1 and 10% of all selected Zeocin resistant transformants), there is a necessity of screening a large number of Zeocin-resistant transformants to locate these valuable clones.
In the previous report on rHuEpo expression in P. pastoris (2), the Lithium chloride transformation method was used to transform yeast cells by the recombinant plasmid, whereas application of electroporation in our study provided higher transformation frequencies as well as higher possibility of obtaining transformants with multi-copies of integrated vector (Easy Select Pichia Expression manual, Invitrogen). Therefore, providing a number of transformed colonies to be screened for their resistance to higher concentration of Zeocin (1 mg/ml), indicated the potential for the presence of multi-copies of expression cassette integrated into the genome (22) and still possibility for selection of the best expression clones by ELISA among a number of screened transformants in small scale expression trials, enabled us to find the highest Epo producing clone.
Moreover, in our investigation the level of Epo expression was improved noticeably through application of a batch culturing strategy by adding acid to maintain the pH at 6.0 (Figure 3B). These results are in line with many previous reports on the importance of the [H+] concentration and specially pH stabilization at 6.0 during yeast induction period for the highest yields of expression of some other heterologous proteins (15, 23 - 26). However, it is important to note that the optimum pH for obtaining the highest level of expression in P. pastoris is a protein-specific factor and greatly depends on physico-chemical properties of the expressed protein that should be determined empirically and as a result, other optimum pH values have been already described for different proteins (27, 28).
Celik et al (2) reported the apparent MW of 32 kDa for P.pastoris expressed rHuEpo on the SDS-PAGE gel which was 2 kDa heavier than the average MW of this protein (30 kDa) as determin
Acknowledgement :
This work was supported by pasteur Institute of Iran and Tehran University of Medical Science "Faculty of Pharmacy".
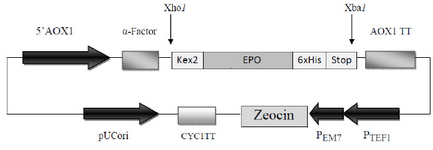
Figure 1. Schematic diagram of recombinant pPICZαA harboring HuEpo gene (PICZαA-Epo: 4.1 Kbp). Xho I and Xba I denote to the restriction sites employed for directional cloning of Epo gene into pPICZαA under control of AOX1 promoter, down stream of secretion signal (α-factor) and cleavage sequence (kex2). AOX1 TT, 6xHis and Stop denote to the transcription termination sequence, His tag and stop codons, respectively
|
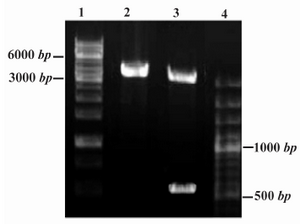
Figure 2. Restriction analysis of PICZαA-Epo construct (4.1 Kbp). DNA fragments were analyzed by 1% agarose gel electrophoresis. Lanes 1 and 4: DNA Ladder SM1163 and SM0321, respectively (Fermentas) Lane 2: Single digestion by Xba I (linear construct, 4.1 Kbp); Lane 3: Double digestion by Xho I and Xba I enzymes (linear pPICZαA and rHuEpo gene are identified as 3.6 Kbp and 540 bp fragments, respectively)
|
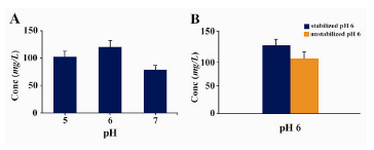
Figure 3. A) Effect of different pH values on expression/ secretion level of rHuEpo. Data are the means ± SD (n=3 each). B) Influence of pH stabilization at 6 throughout the expression time by addition of 0.1 M phosphoric acid in a batch format every 24 hr. Data are the means ± SD (n=3 each, p-value = 0.014). Expression levels in all the trials were measured at the end of induction period (72 hr) by ELISA and Densitometry
|
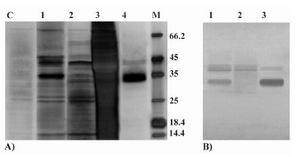
Figure 4. Analyses of P.pastoris-secreted rHuEpo by SDS-PAGE and western blotting. A) Silver stained SDS-PAGE view of different steps in purification of the rHuEpo by IMAC. Lane C: P.pastoris (wild type X-33) transformed with PICZαA plasmid without the inserted Epo gene (control negative); Lane 1: loaded sample (concentrated supernatant); Lane 2: flow through of loading and prewashing; Lane 3: flow through of washing; Lane 4: Eluted (purified) Pichia-derived rHuEpo protein; M: Protein size marker SM0431 (Fermentas). B) Western blot view of crude and concentrated supernatant rHuEpo (Lane 1), flow through of loading and prewashing (Lane 2) and eluted (purified) Pichia-derived rHuEpo protein (Lane 3) using monoclonal anti-human Epo antibody
|
|