Characterization and Functional Assessment of Mouse PPARγ1 Promoter
-
Lachinani, Liana
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology , Isfahan, Iran
-
 Ghaedi, Kamran
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, , +98 311 2612900; kamranghaedi @royaninstitute.org:
Ghaedi, Kamran
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, , +98 311 2612900; kamranghaedi @royaninstitute.org:
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, ACECR, Isfahan, Iran
-
Department of Biology, School of Sciences, University of Isfahan, Isfahan, Iran
-
Tanhaei, Somayeh
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, ACECR, Isfahan, Iran
-
Salamian, Ahmad
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, ACECR, Isfahan, Iran
-
Karamali, Fereshteh
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, ACECR, Isfahan, Iran
-
Kiani-Esfahani, Abbas
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, ACECR, Isfahan, Iran
-
Rabiee, Farzaneh
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, ACECR, Isfahan, Iran
-
Yaghmaei, Marjan
-
Department of Biology, Science and Research Branch, Islamic Azad University, Tehran, Iran
-
Baharvand, Hossein
-
Department of Developmental Biology, University of Science and Culture, ACECR, Tehran, Iran
-
Department of Stem Cells and Developmental Biology, Cell Science Research Center, Royan Institute for Stem Cell Biology and Technology, ACECR, Tehran, Iran
-
Nasr-Esfahani, Mohammad Hossein
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, , +98 311 2612900; mh.nasr-esfahani@royaninstitute.org
-
Department of Cell and Molecular Biology, Cell Science Research Center, Royan Institute for Animal Biotechnology, ACECR, Isfahan, Iran
Abstract: Background: Peroxisome Proliferator Activated Receptor gamma (PPARγ), a member of nuclear receptor superfamily, comprises two isoforms in mouse. These two isoforms are encoded by different mRNAs, which are arisen by alternative promoter usage. There are two promoter regions upstream of PPARγ gene. A 3 kb fragment, containing several transcription factor binding sites, acts as PPARγ1 promoter region. Thus, expression pattern of PPARγ1 isoform is due to the potential transcription factors that could influence its promoter activity. PPARγ, Retinoid X Receptor (RXR) and Vitamin D Receptor (VDR), as nuclear receptors could influence PPARγ gene expression pattern during several differentiation processes. During neural differentiation, PPARγ1 isoform expression reaches to maximal level at neural precursor cell formation.
Methods: A vast computational analysis was carried out to reveal the PPARγ1 promoter region. The putative promoter region was then subcloned upstream of an EGFP reporter gene. Then the functionality of PPARγ1 promoter was assessed in different cell lines.
Results: Results indicated that Rosiglitazone increased PPARγ1 promoter regulated EGFP expression of neural precursor cells during Embryoid Body (EB) formation. Furthermore vitamin D reduced PPARγ1 promoter regulated EGFP expression of neural precursor cells during EB formation through binding to its receptor.
Conclusion: This study suggests that there are potential response elements for PPAR/RXR and VDR/RXR heterodimers in PPARγ1 isoform promoter. Also VDR/RXR heterodimers may decrease PPARγ expression through binding to its promoter.
Introduction :
Peroxisome Proliferator Activated Receptors (PPARs) are ligand-activated transcription factors belong to nuclear hormone receptor superfamily. There are three different isoforms of PPARs: PPARα, PPARβ/δ and PPARγ, encoded by separate genes on different chromosomes. These isoforms exert different functions in the cell and show differential tissue distribution pattern. The main roles of PPARs include cell differentiation, development, and metabolism of macromolecules. The functions of PPARs are mediated through their activation by specific ligands including naturally occurring fatty acids or fatty acid derivatives. To complete this scenario, hetero-dimerization of PPARs with the RXR is required for binding to specific response elements termed: Peroxisome Proliferator Response Element (PPRE) at promoter region of target genes. PPRE consists of hexameric Direct Repeat (DR) pattern (AGGTCA) with a single nucleotide between each hexameric motif. PPAR/retinoid X receptor (RXR) heterodimers bind to these response elements through the PPAR DNA binding domain, which is a highly conserved domain similar to various transcription factors (1,2).
Among PPARs, PPARγ is mainly involved in adipose tissue differentiation and maintenance of adipocyte specific functions. Moreover, it also plays a role in homeostasis of glucose, cholesterol, and insulin sensitivity. Recently, functions of PPARγ in reduction of inflammation, and cell cycle withdrawal have been elucidated (3,4). Besides the mentioned roles, recently we have demonstrated a stage dependent role of PPARγ modulation during neural differentiation of mouse Embryonic Stem Cells (mESC) by retinoic acid treatment (5). Mouse PPARγ consists of two isoforms, PPARγ1 and PPARγ2. Longer isoform (PPARγ2) contains an extra 30 amino acid residues at the amino terminus (6). Both isoforms differ in their expression patterns and tissue distribution. PPARγ1 is mainly distributed in heart, muscle, liver and colon, while, PPARγ2 is highly expressed in the adipose tissue (7).
Productions of these isoforms are under regulation of alternative promoters and different splicing of PPARγ gene. Mouse PPARγ gene comprises 105 kb located at E3-F1 region of chromosome 6.mRNAs of PPARγ1 and PPARγ2 consisting of eight and seven exons, respectively. Six exons are shared in the structure of both PPARγ isoforms. There are extra exons encoding 5’-untranslated regions that are present in the structure of both isoforms. Promoter regions of PPARγ isoforms are distanced 40 kb far from each other, and therefore, they are responsible for different specific expression patterns in several organisms and tissues (6). An intense study of these promoter regions with evaluation of their potential response elements are required to clarify the differential mechanisms of PPARγ isoforms expression. In the present study, we have constructed essential elements of PPARγ1 promoter upstream of EGFP cDNA as a reporter gene to provide a suitable system for evaluation of this region and containing response elements.
Materials and Methods :
Bioinformatics studies: To predict putative promoter regions of mouse PPARγ1 isoform, approximately 200 kb upstream region of PPARγ gene (NC_0000 72.5) was selected for analysis by Genomatix software (http://www.genomatix. de). Furthermore, presence of Transcription Factor Binding Sites (TFBS) in predicted PPARγ1 promoter region was analyzed by several online softwares including Genomatix, TESS (http://www.cbil.upenn. edu), Gene Builder (http://www.itb.cnr.it/sun/ webgene) and TFS EARCH (http://www.cbrc.jp/research/db/TF SEARCH.html). The sequence data are shown in figure 1 and predicted TFBS are demonstrated in table 1.
PCR amplification of PPARγ1 promoter region: DNA was derived from Mouse Embryonic Fibroblast (MEF) cells which were obtained from the Department of Stem cells and Developmental biology (Royan Institute for Stem Cell Biology and Technology) and used as a template in PCR. Specific primers for amplification of predicted PPARγ1 promoter region,-2954 to +178 bp relative to Open Reading Frame (ORF) of PPARγ1, were designed using Oligo6.71 software, introducing VspI and NheI recognition sites at flanking regions of forward and reverse primers, respectively (Table 2), and ordered through Metabion Company (Germany). PCR reactions were performed using ExTaq polymerase (TaKaRa) according to the following protocol: First denaturation was achieved at 94C for 5 min. Amplification reactions were carried out in 35 repetitive cycles during three steps, 45 s at 94C, 45 s at 60C, and 1 min at 72C for denaturation, annealing and extension respectively. Finally, PCR reactions terminated at 72C for 10 min.
Total fragment of PPARγ1 promoter was amplified by Splicing and Overlapping Extension PCR (SOE-PCR) method of four fragments: F1R1 (1190 bp), F2R2 (1040 bp), F3R3 (671 bp) and F4R4 (1032 bp). At first F1R1, F2R2 and F3R3 fragments were amplified and sub-cloned into pTZ57R/T to make one fragment. These fragments demonstrated approximately 100 bp overlapping. The fourth fragment, F4R4, also was amplified and separately sub-cloned into pTZ57R/T. Two constructs were double-digested by XcmI and BamHI and religated at XcmI site to produce total PPARγ1 promoter fragment, F1R4. Finally, total length fragment with VspI-NheI overhangs from pTZ57R/T was sub-cloned into pDB2 target vector (Figure 2).
Amplification of F4R4 fragment (a fragment of about 1 kb) containing a highly GC rich region, was achieved by implementing AMS (Ammonium Sulfate) in reaction to buffer supplemented with 3% DMSO, 0.25 M betaine, 7-deaza dGTP (with 3:1 ratio to normal dGTP).
Plasmid constructions: Amplified fragment of DNA containing PPARγ1 promoter region (3.1 kb) was purified by ethanol precipitation method after gel extraction and inserted into pTZ57R/T vector (Fermentas) using DNA ligation kit (TaKa Ra). Upon blue-white screening of transformed bacterial colonies [DH5α strain of Escherichia coli (E.coli), Fermentas], screening was performed to select those bacterial colonies which contained recombinant plasmid. Thus, positive white colonies were assessed by insert check PCR experiment using T7 and specific promoter primers (Table 2).
Plasmid extraction from positive colonies was carried out using plasmid mini prep kit (Qiagen). Recombinant vectors were sent for sequencing of the insert DNA (Faza Pajouh, Iran). At the next step, PPARγ promoter region was extracted from pTZ57R/T recombinant vectors by VspI and NheI double-digestion and inserted into the same sites inpDB2 vector (kindly provided by Prof. M. Calus, University of Stanford) in place of CMV promoter region and termed pDB2/ PPARγ1.
To construct a promoter free vector, CMV promoter was pulled out from the backbone of pDB2 vector by a VspI-NheI double digestion. VspI-NheI double-digested plasmid backbone was treated by Klenow fragment (Fermentas) at 37 C for 30 min to blunt its sticky ends. Religation was performed to form a circular pDB2 vector without promoter. Finally, recombinant vectors were amplified by transformation in to the DH5α strain of E.coli (Fermentas). Bacterial colonies were checked by PCR insert check analysis.
Cell culture and transfection: CHO-K1 cells were cultured in DMEM/ Ham’s F-12 (Sigma, D8900) medium supplied with 100 U/ml penicillin (Gibco, 15070) under a humidified atmosphere at 5% CO2. CHO cells were plated in density of 1.3x104 cells/cm2. When cells reached to 50-80% of confluency, transfection was carried out by pDB2/PPARγ1 promoter, pDB2 and promoter free pDB2 vectors by lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. After 48 hr of transfection, the cells were fixed by 4% paraformaldehyde/ PBS buffer for 30 min. Meanwhile, nuclei were stained with 4,6 diamidino-2-2- phenylindole (DAPI) for 3 min at room temperature. Green fluorescence of cells was assessed with a fluorescent microscope (Olympus, Japan) and images were taken with an Olympus D70 camera (Olympus, Japan).
Generating stably transformed mouse embryonic stem cells and embryoid body formation and treatments: Mouse embryonic stem cells (mESCs, Royan B1 cells) (8) were cultured in KDMEM (Gibco) with 15% ES-FCS (Gibco), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 2 mM glutamine (Gibco), 0.1 mM non-essential amino acids (Sigma-Aldrich) and 1000 U/ml Leukemia Inhibitory Factor (LIF, Chemicon). mESCs were plated in density of 7.8x104 cells/cm2 in gelatin coated 12-well Tissue Culture Plates (TPP). Transfection was carried out using pDB2/PPARγ1 promoter recombinant vector and lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. After 48 hr of transfection, cells were plated on MTK-Neo feeder cells in the presence of 800 µg/ml of G418 (Sigma). Finally, 24 days later resistant colonies were grown and analyzed by genomic PCR. Stably transformed mESCs were cultured in hanging drops to form Embryoid Bodies (EBs) for two days as previously reported (9). EBs were collected and moved to suspension culture in presence of reduced amounts of serum (10%), 1 µM of retinoic acid (Sigma-Aldrich) and G4 18 (400 µg/ml) for four days.
During the aforementioned 4 days treatment, cells were simultaneously treated with one of the following components: specific PPARγ1 agonists, Rosiglitazone (Cayman Chemical; 5 µM) or selective specific antagonist, GW9662 (Sigma; 10 µM) or Calcitriol (Sigma; 10-8 M). Furthermore, cells were treated by DMSO (10 µl) or ethanol (5 µl) as vehicles for PPARγ agonist, antagonist and Calcitriol (Vitamin D), respectively. On the sixth day, the EBs were collected for real time PCR analysis as previously described (5).
RNA extraction and cDNA synthesis: Total RNAs from transiently transfected CHO-K1 cells and EBs were extracted using RNeasy Mini Kit (Qiagen). Extracted RNAs were treated by DNase I (Fermentas) to remove possible DNA contamination. About
1 µg of total RNA of each sample was used for synthesis of the first strand cDNA using random hexamer primers supplied by Revert Aid First Strand cDNA Synthesis Kit (Fer-mentas).
Real time PCR: Real time PCR reactions were carried out using 5 µl SYBR GreenPCR Master Mix (Takara) and 0.25 pM of specific primers (Table 2), 25 ng of cDNA in total volume of
10 µl. All reactions were held in triplicates and normalized by GAPDH. All data’s were analyzed by ∆∆Ct method.
Flow cytometry: To quantify the fluorescence intensity of EGFP, transiently transfected CHO-K1 cells were detached by Trypsin/EDTA (Gibco),
48 hr post-transfection and analyzed by Becton Dickinson FACS Calibure flow cytometer (USA) as follows: for each sample, 104 events were recorded in the forward light scatter/side light scatter (FSC/SSC) dot plot. Then a gate was used to select single cells from aggregated and debris.
Green fluorescence of EGFP was detected in the fluorescence detector 1 (FL-1) with a 530/30 nm band pass filter. Data obtained from flow cytometer instrument were analyzed by using Cell-Quest Pro and WinMDI 2.9 software. To reduce the transfection efficiency effect, this experiment was done three times independently and the average of fluorescence intensity was calculated and considered for analysis.
Statistical analyses: Data were expressed as means±SEM obtained from three independent replicates of observations. Differences between the expression patterns of the samples were determined using student’s t-test and were judged to be significant at p<0.01 and 0.05.
Results :
Bioinformatics studies of PPARγ1 promoter region and amplification of target region: Based on bioinformatics studies, six potential promoter upstream regions of PPARγ gene were predicted. According to transcription initiation site only one of these potential regions was able to be used for encoding PPARγ1 mRNA. This region was similar to the previously determined PPARγ1 promoter region with little differences in several nucleotides. Thus, considering the previous studies (6), 3.1 kb DNA fragment was selected and analyzed for presence of possible TFBS (Figure 1A). Data predicted presence of several putative TATA boxes (Table 1). This fragment was characterized as a highly GC rich fragment (about 1 kb) with >80% CG content and 332 bp of CpG island (Figure 1B). According to bioinformatics results from Genomatix, TESS, GeneBuilder and TFSEARCH softwares, different TFBSs were predicted at this promoter region (Table 2). Among the predicted sequences, there were response elements for VDR-RXR and PPAR-RXR heterodimers and PPARγ homodimer binding sites (Figure 1C).
PPARγ1 promoter region subcloning and promoter activity confirmation: PPARγ1 promoter regions were successfully amplified as F1R1 (1190 bp), F2R2 (1040 bp), F3R3 (671 bp) and F4R4 (1032 bp) (Figure 3). As described in material and methods, F1R1, F2R2 and F3R3 fragments were amplified and sub-cloned into pTZ57R/T to make one fragment. The fourth fragment, F4R4, also was amplified and separately sub-cloned into pTZ57R/T.
At the next step, whole of the PPARγ1 promoter region was successfully amplified and cloned into pDB2 target vector. CMV promoter of pDB2 vector was removed by VspI and NheI digestion and PPARγ1 promoter region was replaced at corresponding sites. Thus, in pDB2/PPARγ1 promoter recombinant vector, expression of EGFP reporter gene was under regulation of PPARγ1 promoter region (Figure 2). To confirm PPARγ1 promoter activity, pDB2-PPARγ1 promoter recombinant vector was transfected into CHO-K1 cells. After 48 hr of transfection, green fluorescence was observed in cells and confirmed promoter activity and functionality of recombinant vector (Figures 4E and 4H). Simultaneously, original vector of pDB2, containing CMV promoter and promoter-lacking pDB2 vector were transfected into CHO cells. Cells transfected by original pDB2 vector expressed EGFP at high levels (Figures 4A, 4D and 4G) when compared with untransfected cells (Figures 4C, 4F and 4I). Whereas, promoter-lacking pDB2 vector transfection had no EGFP expression result (data not shown).
To evaluate and compare EGFP expression levels under control of PPARγ1 promoter, flow cytometric analysis was performed. Data showed a reduced EGFP expression pattern under control of PPARγ1 promoter relative to viral CMV promoter. These data implicated that relative activity of PPARγ1 promoter is about 0.2 fold of CMV promoter (Figure 4J).
Furthermore, to evaluate the functionality of PPARγ1 promoter in another cell line, stably transformed mESCs with the recombinant vector (pDB2-PPARγ1 promoter) was implemented for analysis of the EGFP expression level. Real time PCR analysis revealed significant difference in EGFP expression level in stably transformed mESCs compared to the untransfected mESCs (Figure 4K).
As several response elements for VDR-RXR and PPAR-RXR heterodimers and PPARγ homodimer binding sites were identified in putative PPARγ1 promoter region, the effects of PPARγ agonist (Rosiglitazone), PPARγ antagonist (GW9662) and vitamin D (Calcitriol) on promoter activity of PPARγ1 promoter were assessed using real time PCR analyses for EGFP expression. We have already shown that 5 µM of Rosiglitasone and 10 µM of GW 9662 caused activation and inactivation of PPARγ, respectively as nuclear localization of PPARγ increased upon activation and decreased during inactivation (5,10). Our results indicated that Rosiglitazone increased PPARγ1 promoter regulated EGFP expression of neural precursor cells during EB formation (Figure 4L). Thus, it seems that active heterodimers of PPARγ/RXR interact with PPARγ1 promoter region and this region contains potential response elements for PPARγ/RXR heterodimers. On other hand, GW9662, potent antagonist of PPARγ, reduced EGFP expression in these cells (Figure 4L).
Due to the recently published function of vitamin D in nervous system, the effect of vitamin D was examined on EGFP expression in stably transformed mESCs that underwent neural precursor cell formation. Data revealed vitamin D reduced PPARγ1 promoter regulated EGFP expression of neural precursor cells during EB formation through binding to its receptor (Figure 4L).
Discussion :
PPARs are members of the nuclear-receptor superfamily of proteins and act as nuclear transcription factors where they form a heterodimer with the Retinoid X Receptor (RXR). The PPAR-RXR heterodimer binds to PPAR Response Elements (PPRE) in the promoter of PPAR-responsive genes (1,3,11). Among various PPARs, PPARγ consists of two different isoforms (PPARγl and PPARγ2) because of alternative processing of PPARγ mRNA.
In this manuscript, cloning of PPARγ1 promoter is reported and it subfunctionality through construction of a vector regulating EGFP expression is approved. However, the strength of PPARγ1 was estimated to be 20% of the CMV promoter. Bioinformatics studies revealed presence of different transcription factor-response elements at promoter region of mouse PPARγ gene. The functionality of response elements for PPAR/RXR and VDR/ RXR at promoter region of mouse PPARγ1 isoform was pinpointed. PPARγ and vitamin D activated receptor exert their activity through binding to specific DNA sequences at promoter region of target genes. Their binding to specific response elements requires hetero-dimerization with RXR (3,12).
PPARγ is a transcription factor mainly expressed in adipose tissues. Our recent studies suggest that PPARγ also maintains a role in neural differentiation. During neural differentiation of mouse embryonic stem cells, expression of PPARγ1 isoform is raised and reaches maximal level during neural precursor cell formation (5). Treatment of mESC by retinoic acid during this procedure causes activation of RXR. In this stage Rosiglitazone, a synthetic PPARγ agonist, activates PPARγ and induces PPAR/RXR heterodimer formation. Subsequently, activated PPAR/RXR heterodimers could bind to PPREs in promoter regions of target genes.
Based on bioinformatics studies, there are predicted response elements of PPARγ/RXR and PPARγ at PPARγ1 promoter region, respectively at -577 and -254 bp of PPARγ1 ORF. In this study, treatment of stably transfected mESCs by GW9662, a PPARγ antagonist decreased PPARγ promoter activity to 0.5 fold of untreated cells. In addition, Rosiglitazone treatment caused an increase in PPARγ promoter activity. This data suggest that PPARγ/RXR heterodimers could regulate PPARγ expression by binding to respective response element at PPARγ promoter.
Vitamin D, as a nuclear hormone and its nuclear receptor (Vitamin D receptor) regulate transcription of several genes in neurons and neuronal precursor cells (13,14). This nuclear receptor associates with vitamin D and forms heterodimers with RXR and exerts its activity through binding to vitamin D response elements of target genes. In adipocyte, vitamin D and VDR inhibit both PPARγ activity and adipogenesis (15).
Conclusion :
Based on bioinformatics studies on PPARγ1 promoter region, a potential VDR/ RXR heterodimer response element was predicted at -149 bp of PPARγ1 ORF. PPARγ1 promoter activity in stably transfected mESCs after treatment by vitamin D was decreased to 0.7 fold of untreated cells at day 6 of EBs, when both PPARγ and RXR were transcriptionally active. We hypothesized that VDR/ RXR heterodimers may decrease PPARγ expression through binding to its promoter. Clearly, more work is needed to develop a comprehensive understanding of the cellular and molecular mechanisms in regulating PPARγ expression by vitamin D.
Acknowledgement :
This study was supported by a grant from Royan Institute awarded to Kamran Ghaedi.
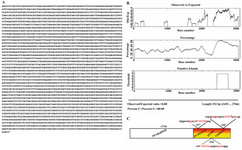
Figure 1. Sequence of mouse PPARγ putative core-promoter. A) PPARγ1 promoter region sequence. B) CpG plot of PPARγ1 core-promoter region (EMBL-EBI: http://www.ebi.ac.uk/Tools/emboss/cpgplot/index.html). C) Diagram of GC rich region of PPARγ1 promoter region and predicted response elements on it
|

Figure 2. Schematic representation of PCR amplification of PPARγ1 promoter region by SOE-PCR and subcloning into pDB2 reporter vector
|
![Figure 3. PCR steps for constructing different PPARγ1 promoter region. PCR-product bands: the molecular size marker [100 bp; Fermentas] (M) and four fragments of PPARγ1 promoter region; F1R1 (1190 bp), F2R2 (1040 bp), F3R3 (671 bp) and F4R4 (1032 bp) are indicated by arrow heads. Stars indicate nonspecific bands](Images/Articles/95/f3_small.png)
Figure 3. PCR steps for constructing different PPARγ1 promoter region. PCR-product bands: the molecular size marker [100 bp; Fermentas] (M) and four fragments of PPARγ1 promoter region; F1R1 (1190 bp), F2R2 (1040 bp), F3R3 (671 bp) and F4R4 (1032 bp) are indicated by arrow heads. Stars indicate nonspecific bands
|

Figure 4. Assessment of the functional activity of PPARγ1 promoter. Transiently transfected CHO cells by pDB2 vector (A, D, G) and pDB2-PPARγ1 promoter vector (B, E, H) and untransfected cells (C, F, I). As shown in this figure, the cloned fragment was bona fide part of PPARγ1 promoter region with a weaker activity than CMV promoter. Nuclei counterstaining with DAPI (upper panel), EGFP fluorescence (middle panel), merged figures (lower panel) are shown. J) Comparison of PPARγ1 promoter activity with CMV promoter using flow cytometry as indicated PPARγ1 promoter is a weaker promoter than CMV. K) EGFP expression level in stably transformed mESCs by pDB2-PPARγ1 promoter vector compared with untransfected cells showing functional activity for PPARγ1 promoter. L) Treatment of stably transfected mESCs by Rosiglitasone (Rosi: 5 µM) or GW9662 (GW: 10 µM) or Calcitriol (VD:10-8 M) as described in materials and methods. As predicted at this promoter region (Table 2), there were response elements for VDR-RXR and PPAR-RXR heterodimers and PPARγ homodimer binding sites (Figure 1C). EGFP expression levels in these cells were compared with untreated stably transformed mESCs by pDB2-PPARγ1 promoter by real time PCR. In this study, treatment of stably transfected mESCs by Rosiglitasone and GW9662, increased and decreased PPARγ promoter activity 1.5 and 0.5 fold, respectively. Moreover, vitamin D reduced PPARγ1 promoter regulated EGFP expression in neural precursor cells (approximately 40%). Scale bar is 200 µm
|
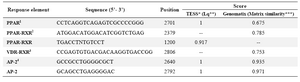
Table 1. Predicted transcription factor binding sites for mouse PPARγ1 promoter
*TESS: Transcription element search software on the WWW.
**Lq: The ratio of La / L_M, where L_M is the maximum La possible for the site model. The best score is 1.0. Thus La/ is the ratio of log-likelihood score to the length of the site. The best score for La/ is 2.0. For further information please see the following site: http://www.cbil.upenn.edu/cgi-bin/tess/tess?RQ=MRZ-leg&job=W0502026399&is=1&nr=50&att=beg&fr=0&mask=-1.
***Matrix similarity: The matrix similarity is calculated as described in http://www.genomatix.de/online_help/help/scores.html?s=b66803c222e3ce9257cd2e748b244230#msim.
A perfect match to the matrix gets a score of 1.00 (each sequence position corresponds to the highest conserved nucleotide at that position in the matrix), a "good" match to the matrix usually has a similarity of >0.80.
1 Peroxisome proliferator-activated receptor
2 PPAR heterodimer with retinoid X receptor
3 Vitamin D receptor heterodimer with retinoid X receptor
4 Activator protein 2
|

Table 2. List of primers used in this study
F and R, are referred as forward and reverse primers, respectively. Restriction sites are underlined
|
|