Expression of the Hepatitis C Virus core-NS3 Fusion Protein on the Surface of Bacterial Ghosts: Prospects for Vaccine Production
-
Tayebinia , Minoosadat
-
Diagnostic Laboratory Sciences and Technology Research Center, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
 Sharifzadeh , Sedigheh
Diagnosis Laboratory Sciences and Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran, Email: sharifsd@sums.ac.ir
Sharifzadeh , Sedigheh
Diagnosis Laboratory Sciences and Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran, Email: sharifsd@sums.ac.ir
-
Division of Medical Biotechnology, Department of Medical Laboratory Sciences, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Rafiei Dehbidi , Gholamreza
-
Diagnostic Laboratory Sciences and Technology Research Center, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Zare, Farahnaz
-
Diagnostic Laboratory Sciences and Technology Research Center, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Ranjbaran, Reza
-
Diagnostic Laboratory Sciences and Technology Research Center, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Rahimi, Amir
-
Bioinfirmatic and Computational Biology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
-
Miri, Mohammad Reza
-
The Persian Gulf Marine Biotechnology Research Center, the Persian Gulf Biomedical Sciences Research Institute, Bushehr University of Medical Sciences, Bushehr, Iran
-
Behzad-Behbahani, Abbas
Diagnosis Laboratory Sciences and Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran, Tel: +98 71 32270301; Fax: +98 71 32270301; E-mail: behzadba@sums.ac.ir, behzadba@gmail.com
-
Division of Medical Biotechnology, Department of Medical Laboratory Sciences, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
Abstract: Background: Antigen presentation using bacterial surface display systems, on one hand, has the benefits of bacterial carriers, including low-cost production and ease of manipulation. On the other hand, the bacteria can help in stimulating the immune system as an adjuvant. For example, using bacterial surface display technology, we developed a hepatitis C virus (HCV) multiple antigens displaying bacteria's surface and then turned it into a bacterial ghost.
Methods: The HCV core and NS3 proteins' conserved epitopes were cloned into the AIDA gene plasmid as an auto transporter. The recombinant plasmid was then transformed into Escherichia coli (E. coli) Bl21 (DE3). Recombinant bacteria were then turned into a bacterial ghost, an empty cell envelope. Whole-cell ELISA, flow cytometry, and Western blot techniques were used for monitoring the expression of proteins on the surface of bacteria.
Results: A fusion protein of HCV core-NS3-AIDA was successfully expressed on the E. coli Bl21 (DE3) surface and confirmed by western blotting, Enzyme-Linked Immunosorbent Assay (ELISA), and flow cytometry detection techniques.
Conclusion: The presence of HCV antigens on non-pathogen bacteria surfaces holds promise for developing safe and cost-benefit-accessible vaccines with optimal intrinsic adjuvant effects and exposure of heterologous antigens to the immune system.
Introduction :
Hepatitis C infection is a serious chronic disease caused by the hepatitis C virus (HCV). The viral infection spreads to an uninfected person when contaminated blood enters that person’s bloodstream. HCV has excessive genetic variation resulting from defects in RNA-dependent RNA polymerase repair activity due to genetic polymorphism. Accordingly, there are several unique genotypes of HCV around the world. So far, eight distinct genotypes and more than 86 subtypes are known 1.
At present, PEGylated Interferon (PEG-IFN) and Ribavirin comprise antiviral therapy, which is both expensive and, therefore, often out of reach for many people. In addition to economic factors, it is often not sufficiently effective in all patients while also causing undesirable side effects 2. Consequently, vaccination is the most effective strategy to prevent and control such infectious diseases 3.
HCV is a positive single-stranded RNA with an envelope that belongs to the Flaviviridae family. Its’ genome consists of about 1600 nucleotides that encode a polyprotein with 3010 amino acids. There are two types of HCV proteins encoded by the genome: structural (Core, E1, and E2) and non-structural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) 4. Several studies suggest that cellular and humoral immune responses play an important role in the clearance of HCV infection 5,6. Given this, a vaccine's design to induce both cellular and humoral responses to HCV infection seems desirable 7. HCV core proteins appear to elicit the broadest range of humoral and cellular immune reactions across virus variants. A combination of CD4+ and CD8+ responses can effectively control HCV infection 8. Non-structural protein NS3 that contains a helicase in the C-terminal region and a protease in the N-terminus is regarded as the preferred target for natural or therapeutic eradication of the virus 9.
Despite the prevalence of HCV and its serious effects on health, no effective vaccine has been introduced. Numerous techniques used to increase the immunogenicity of multi-epitope peptide vaccines consist of using the proper adjuvant and enhancing the delivery system 10,11.
The microbial cell-surface display is a technique used for expressing proteins or peptides on the surface of bacteria by binding them with the anchoring features or motifs of the cell surface. The protein of interest can be transported from the cytoplasm to the bacteria's surface using this technique. In this method, the protein of interest (passenger protein) is fused to a carrier protein and exported from the cytoplasm to the surface 12-14. A utility of these strategies is in developing live vaccines by displaying antigenic proteins at the surface of micro-organisms. This kind of vaccine has some advantages over traditional vaccines, like the antigens expressed in this way are recognized by the immune system 15-17.
Auto display of epitopes could, by extrapolation, result in enhanced immune system stimulation and could be used for vaccine development. In addition, utilizing non-pathogenic bacterial cells to display different epitopes has been shown to elicit antigen-specific antibody responses 18-20.
Bacterial Ghosts (BG) are empty cell envelopes originating from Gram-negative bacteria. BG has natural adjuvant properties due to its nature as bacterial envelope complexes. A BG can stimulate the immune system without any additional adjuvants 21. In the present study, using Adhesin Involved in Diffuse Adherence (AIDA) autotransporter protein and BG technology, multiple conserved epitopes of HCV Core and NS3 proteins were displayed on the surface of Escherichia coli (E. coli) as an antigen carrier and strong adjuvant for the immunization.
Materials and Methods :
Bacterial strains, reagents, and culture media: Cloning and plasmid extraction methods were performed using E. coli strain DH5a. E. coli strain BL21 (DE3) was used as a host cell to express foreign proteins on the surface of bacteria. The restriction enzymes (SmaI and XbaI) were obtained from JenaBioscience (Germany). Pfu DNA polymerase, T4 DNA ligase, and Page Ruler™ Prestained Protein Ladder were purchased from Thermo Fisher Scientific (Grand Island, NY). Primers were ordered from Macrogen (South Korea). Gel and plasmid extraction kits were obtained from Qiagen (Hilden, Germany). Ampicillin was obtained from Sigma-Aldrich (USA). Anti-Hepatitis C Virus Core 1b antibodies [C7-50] (ab2740) were purchased from Abcam. The HCV BLOT 3.0 kit was purchased from MP Biomedicals. Luria-Bertani (LB) medium was used for the growth and maintenance of E. coli strains. Surface display experiments were conducted in a minimal sal medium 22 with a sterile glucose solution (500 g l-1) as a carbon supply.
Construction of recombinant AIDA-HCV Core/Ns3 plasmid: The plasmid pMK90 used for surface expression was kindly provided by Dr. I. Benz, (University of Münster, Germany) 23. It is a derivative of pBR322 containing an ampicillin resistance gene under the control of the AIDA promoter. The plasmid consisted of a signal peptide of 49 amino acids, followed by a linker region of 78 amino acids with multiple cloning sites for a recombinant passenger protein followed by the AIDA-I translocation unit (AIDAc). The DNA fragments encoding HCV Core and NS3 genes were fused using overlap-extension PCR (OE- PCR) as in previous work 21. The chimeric fragment (Core-NS3) was then amplified by Pfu DNA polymerase using a forward oligonucleotide primer (coreF1) (5'-CCCACAGGACGTCAA GTTC-3') and reverse primer (NS3R1) (5'-CATCTAG AGTGACGACCTCCAGGTCGG -3') at concentrations and thermal cycling program as previously described 21. Following pMK90 digestion by XbaI and SmaI, the chimeric fragment (HCV Core-NS3) was inserted into the vector's multiple cloning site (pMK90HCV), and the vector was transformed into E. coli BL21 (DE3) strain using CaCl2 method.
The bacteria culture selected the correct cloned gene on LB agar containing ampicillin (100 mg l-1). According to the manufacturer's instructions, plasmids were then extracted from the transformed bacteria using a plasmid extraction kit (Qiagen, USA). To verify correct nucleotide sequences and gene orientation, restriction enzyme analysis, colony PCR.
Bacterial production with displaying of HCV Core/Ns3 antigen on the surface: E. coli BL21 (DE3) transformed with the corresponding pMK90 HCV (test), and pMK90 (control) derivative was separately cultured at 37°C in shake flasks with 250 ml Luria-Bertani (LB) medium supplemented with 100 μg/ml ampicillin (Amp). After 6-8 hr incubation for the growing bacteria and gene expression at OD 660 nm=2.5, the cells were pelleted by centrifugation (5000 rpm) at 4°C for 5 min and washed with cold Phosphate-Buffered Saline (PBS).
Bacterial ghost production: A phage-derived lytic protein E was expressed in E. coli to produce BGs, as previously described 21.
Whole-cell Elisa: To investigate the presence of HCV core-NS3 antigens on the surface of transformed E. coli BL21 (DE3), the whole-cell ELISA method was performed 24. Briefly, 100 µl of bacterial suspension in PBS (phosphate-buffered saline, pH~7.4) was added to microplate wells and incubated at 37°C overnight. The wells were then blocked using 200 µl of 3% (w/v) skimmed milk in PBS for 1 hr incubation at 37°C. After 4 times washing with 350 µl PBS, 100 µl (1:1000 in PBS) of anti-HCV core 1b antibody (Abcam, USA) was added to each well and incubated at 37°C for 1 hr. Following 3 times of washing with 350 ml PBS, 100 ml (1:1000) PBS was added to each well, incubated at 37°C for 1 hr, followed by the addition of goat anti-mouse antibodies. After washing three times with PBS, 50 µl of 3,3′,5,5′-tetramethylbenzidine (TMB) as substrate was added to each well and incubated in the dark for 15 min. To stop the reaction, 50 µl of 2 M sulfuric acid was added to each well. The absorbance of the wells was measured at 405 nm for 15 and 30 min.
Isolation and purification of the outer membrane protein (OMP): The OMP was obtained as previously described with some modification 22. Briefly, the cells of 250 ml culture were harvested by centrifugation, washed with buffer A (50 mm Tris/HCl pH=7.5), and disrupted by sonication (6×3 s, frequency of 30 kHz). After ultracentrifugation (36000 g for 40 min), the whole membrane fractions were collected and washed with buffer A. After washing with 0.1% sarcosyl in buffer A for 1 hr at 4°C, the membrane pellet was collected by ultracentrifugation. The protein fractions were obtained in the remaining pellet by dissolving in 7.5 ml of buffer A contained 5 mM EDTA and 1% (v/v) Triton X-100. The resultant membrane protein samples were stored at -20°C before analysis.
SDS-PAGE and western blot analysis: SDS-PAGE and western blot analysis identified fractions containing OMP. Briefly, the samples loaded directly or after incubation at 100°C for 5 min after mixing with loading buffer on 12% acrylamide gels. Using Coomassie Brilliant Blue, the bands of protein were visualized. Proteins were transferred from polyacrylamide gels to PVDF membrane (Amersham, USA) by the semi-dry blot technique (Trans-Blot® SD Semi-Dry Transfer Cell, Bio-Rad). The transfer was achieved at a current of 0.8 mA/cm2 for 1 hr at room temperature. The membrane was blocked by incubation in PBS with 5% milk powder at 4°C after the transfer of proteins. According to the manufacturer's instructions the remaining steps were accomplished based on the MP Diagnostics HCV BLOT 3.0 kit.
Flow cytometry assay: E. coli BL21 (DE3) harboring pMK90HCV (test) and pMK90 (control) were used for the experiment. Briefly, E. coli was incubated overnight at 37°C in LB/Amp broth and then sub-cultured in fresh LB/Amp medium for approximately an hour. The subculture was grown to reach 4×108 CFU/ml, with an optical density between 0.5 and 0.6 at 600 nm (path length 1 cm). Aerated orbital shaking at 200 rev min-1 was used to aerate all broth cultures grown at 37°C. The cells were treated with 100 μl of a solution containing anti-hepatitis C Virus Core 1b monoclonal antibody (diluted 1:100 in PBS) and incubated for 1 hr at room temperature. Cells were washed three times with 500 μl of PBS. The second incubation step was conducted with 100 μl of goat anti-mouse IgG conjugated with FITC (diluted 1:50 in PBS) and the cells were incubated for 1 hr at room temperature in a dark place. Following three times of washing with PBS, the cell pellet was suspended in 500 µl of PBS and analyzed by flow cytometry (Becton Dickinson, Sydney, Australia).
Results :
Construction, identification, and sequencing of recombinant AIDA-HCV Core/Ns3 plasmid : After enzymatic digestion with SmaⅠ and XbaⅠ, Core/NS3 fragment in 585 bp length and pMK90 liner fragment containing AIDA fragment in 1320 bp were obtainable in prediction (Figure 1). Sanger sequencing was performed to verify recombinant plasmid. The vector harboured a natural AIDA promoter, AIDA signal sequence, Core-NS3 in the passenger part, a linker region, and finally, the translocation unit (AIDAC) sequence. No mutation was observed due to the misincorporation of DNA polymerase. The resultant Core-NS3 fusion protein consists of 585 bp (accession number: GenBank ON128514), which was equivalent to 195 aa with a molecular mass of 21.4 kDa. Cleavage of the N-terminal signal peptide results in a protein of 560 amino acids (74.87 kDa) (Figure 2).
Expression of HCV Core-NS3 fusion protein on the surface of E. coli: To determine the potential role of AIDA transmembrane protein to transport HCV Core/NS3 antigens on the surface of bacteria, E. coli BL21 (DE3) as an OmpT-negative strain was transformed with the surface display vector pMK90HCV (Figure 2). Recombinant E. coli harboring HCV Core/NS3 sequences were cultivated, and the expression of proteins on the surface of the cells was analyzed by SDS-PAGE and western blotting (Figure 3). The HCV core/NS3 linked to the AIDA protein was detected in both the lysate and outer membrane fractions of E. coli cells harboring pMK90HCV. Overall, the chimeric Core/NS3-AIDAC protein's molecular weight was approximately 75 kDa (i.e., 21.4 kDa for Core-NS3 fusion protein and 53.5 kDa for AIDAC protein, respectively).
Production of bacterial ghosts: A lysis protein E was expressed in E. coli BL21 (DE3) to produce BG. The bacterial cells were then cultured below the recommended temperature (28°C/ control) to suppress the lytic gene activity. A temperature increase of 42°C triggered the activation of protein E for 30 min before adding 0.2 M MgSO4. The entire process was completed in around 90 min. By adding antibiotics (1% Pen-Strep (Invitrogen), the bacteria that were not able to become BG were inactivated.
The BG was centrifuged and cultured in LB medium contains antibiotic chloramphenicol (33 g/ml) to determine the purity of the BG. During 48 hr, no live bacteria were detected (colonies).
Whole-cell ELISA assay: Using the optimal E. coli concentration, the expression level of HCV Core/NS3epitopes on the bacterial surface of E. coli BL21 (DE3)/pMK90HCV was determined by whole-cell ELISA. E. coli BL21 (DE3)/ pMK90 and E. coli BL21 (DE3) were used as the negative controls. E. coli BL21 (DE3)/pMK90 and E. coli BL21 (DE3) were coated in an ELISA plate. The Core/NS3 surface-exposed protein was detected by using the anti-hepatitis C virus core 1b antibody. A strong yellow color formed in wells containing the BL21 (DE3)/pMK90HCV confirmed the presence of Core/NS3 epitopes on the surface of bacteria by the findings from the Western blot analysis (Figure 4).
Evaluation of the surface display by FACS analysis: To study HCV antigens, display efficiency on the bacterial surface both transformed bacteria with pMK90HCV (test group) and pMK90 (control group) were labeled with primary anti-HCV Core 1b mouse monoclonal antibody coupled to secondary goat anti-mouse IgG-FITC antibody. Data analysis indicated that the fluorescence intensity of bacteria expressing the Core/Ns3 fusion protein was shifted to a higher intensity than the control cells treated as the test group (Figure 5). Results indicate the E. coli cells express fusion HCV Core/NS3 protein on their surface.
Discussion :
Vaccination is a particularly effective method of preventing certain infectious diseases. However, the efficient delivery of antigens is the major goal of vaccination. Therefore, bacterial vectors as a biological delivery system are of interest among the various methods used to transfer a protein as an effective antigen to the cells.
BG have the potential to become novel vaccine platforms. Biologically speaking, these systems might be effective and well-compatible for turning drugs, enzymes, and genetic materials into tissues, organs, and cells. Further, its intact surface structure makes it a valuable adjuvant and targeting vehicle. Besides, the bacterial surface display can be used as vaccine delivery, and there are many advantages to using antigen surface display technology to deliver antigens. For example, using bacterial surface display technology and using a bacterial autotransporter system 17, we developed a hepatitis C virus (HCV) multiple antigens displaying on bacteria's surface.
The Core protein sequence in the HCV virus is one of the most conserved parts in the virus's genome of various genotypes. Several epitopes have been identified by B and T cells in the core protein of HCV, and serum antibodies against core immunogenic epitopes included amino acids 7-31, 21-49, 45-63, and 99-113 25. In addition, the amino acids 18-181 were selected from a core protein, with epitopes for CTL in different HLAs, 11 epitopes for antibody production in mice and humans, and 11 epitopes for helper T-cells.
The significant challenge for exporting a protein to the bacterial surface is translocating the desired gene product from a cell periplasm to the external cell surface. In the present study, we employed the AIDA gene, one of the autotransporter proteins whose primary sequence contains all the information needed to cross target protein to the outer membrane of bacteria. The N terminus of AIDA contains a signal peptide responsible for exporting the target protein through the inner membrane, followed by the AIDA gene's passenger sequence within the PMK90 vector. The passenger sequence was removed by restriction enzymes and replaced by the target sequence. Finally, the C-terminal of the AIDA gene contains the translocator sequence responsible for displaying the target protein on the Bl21 (DE3) bacterium's surface.
Due to the location of the core and NS3 regions within the genome which contain epitopes that facilitate a cell-mediated immune response, as well as a humoral immune response. As an alternative to restriction digestion and ligation, two HCV Core and NS3 genes were attached by PCR SOEing 26. The chimeric product did not exhibit nucleotide misincorporation, as indicated by DNA analysis. The orientation of the core-NS3 sequence was designed from 5' to 3' because the 5' region is located outward the outer membrane lipid and readily exposes to the immune system and stimulates antibody production.
By whole-bacterial cell ELISA assay, we found that marked reactivity with anti‐HCV core 1b antibody, suggesting that the HCV proteins on the bacterial surface could fold into the three-dimensional structure in such a way that at least the linear epitopes were exposed on the protein surface. The results were further supported by Western blot and flow cytometry analysis. When immunoreactivities of the recombinant E. coli were tested using five different HCV patients' sera (data not presented), all reacted with the surface antigens compared with the control group (non-modified bacteria).
Recently, the E. coli BL21 (DE3) bacterial surface display system was used to express rotavirus virus protein 8 (VP8*) as a ligand to detect Histo-Blood Group Antigens (HBGAs) receptors, which play an important role in rotavirus infection. Western blot and ELISA evaluated the function of the bacterial cell surface display system as well 27.
Conclusion :
Our analyses of cell surface display systems and producing BGs using HCV core-NS3 proteins indicated that the technology might be useful in producing a whole-cell killed vaccine harboring HCV antigens. Since E. coli, as gram-negative bacteria, contain LPS, it acts as a potent adjuvant in vaccine administration. However, this technology can be applied to any bacterium for different applications.
Acknowledgement :
We would like to thank the staff of the Diagnostic Laboratory Sciences and Technology Research Center, Shiraz University of Medical Sciences for their technical assistance. The Ethics Commission of Shiraz University of Medical Sciences approved this study (IR.SUMS.REC.1394.5256).
Funding: This work is supported by Shiraz University of Medical Sciences, Shiraz, Iran (Project No.93-01-10 9022). The authors report no involvement in the research by the sponsor that could have influenced the outcome of this work.
Conflict of Interest :
None. The authors certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.

Figure 1. Electrophoresis of the enzymatic digest of the PMK90 plasmid vector using the restriction enzymes SmaI and XbaI. A) plasmid vector digested with SmaI, B) plasmid vector digested with XbaI, C) gel electrophoresis of the chimeric core-NS3 fragment digested with XbaI.
|
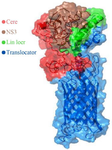
Figure 2. Schematic model of the HCV core-NS3 and AIDA translocator protein on the surface of E. coli.
|
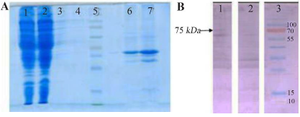
Figure 3. SDS-PAGE and Western blot assay on proteins obtained from various fragments of genetically engineered bacteria and control separated by ultracentrifugation.
A) SDS-PAGE on various fragments separated by ultracentrifugation. 1: Cytoplasmic proteins of coli BL21(DE3) strain transfected with pMK90 vector, 2: Cytoplasmic proteins of E.coli BL21(DE3) strain transfected with pMK90-HCV vector, 3: Inner proteins of E.coli BL21(DE3) strain transfected with pMK90 vector, 4: Inner proteins of E.coli BL21(DE3) strain transfected with pMK90-HCV vector, 5: Protein size marker, 6: Outer membrane proteins of E. coli BL21(DE3) strain transfected with pMK90 vector, 7: Outer membrane proteins of E. coli BL21(DE3) strain transfected with pMK90-HCV vector. B) Western blot of the outer membrane protein of E. coli using a human serum that contains high titer antibodies to HCV. Lane 1: OM fraction of E. coli BL21 (DE3)+pMK90HCV, Lane 2: OM fraction of E. coli BL21(DE3) pMK90(Negative control), Lane 3: Protein size marker.
|
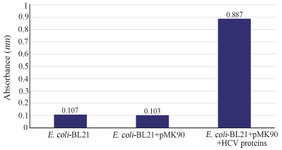
Figure 4. Detection of HCV Core/NS3 fusion protein on the surface of E. coli BL21(DE3) by ELISA.
|
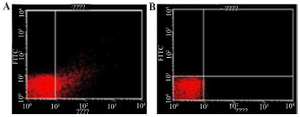
Figure 5. Cells analyzed by flow cytometry anti-hepatitis C Virus Core 1b monoclonal antibody linked to goat Anti-mouse using IgG-FITC secondary antibody.
A) coli (pMK90HCV), B) E. coli (pMK90) as negative control.
|
|