Comparison of Proliferative and Multilineage Differentiation Potential of Sheep Mesenchymal Stem Cells Derived from Bone Marrow, Liver, and Adipose Tissue
-
Heidari, Banafsheh
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
 Shirazi, Abolfazl
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, +98 21 22432020; shiraziabbas@yahoo.com
Shirazi, Abolfazl
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, +98 21 22432020; shiraziabbas@yahoo.com
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Department of Gametes and Cloning, Research Institute of Animal Embryo Technology, Shahrekord University, Shahrekord, Iran
-
Akhondi, Mohammad Mehdi
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Hassanpour, Hossein
-
Department of Gametes and Cloning, Research Institute of Animal Embryo Technology, Shahrekord University, Shahrekord, Iran
-
Behzadi, Bahareh
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Naderi, Mohammad Mehdi
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Sarvari, Ali
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Borjian, Sara
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
Abstract: Background: Despite major progress in our general knowledge related to the application of adult stem cells, finding alternative sources for bone marrow Mesenchymal Stem Cells (MSCs) has remained to be challenged. In this study successful isolation, multilineage differentiation, and proliferation potentials of sheep MSCs derived from bone marrow, adipose tissue, and liver were widely investigated.
Methods: The primary cell cultures were prepared form tissue samples obtained from sheep 30-35 day fetus. Passage-3 cells were plated either at varying cell densities or different serum concentrations for a week. The Population Doubling Time (PDT), growth curves, and Colony Forming Unit (CFU) of MSCs was determined. The stemness and trilineage differentiation potential of MSCs were analyzed by using molecullar and cytochemical staining approaches. The data was analyzed through one way ANOVA using SigmaStat (ver.2).
Results: The highest PDT and lowest CFU were observed in adipose tissue group compared with other groups (p<0.001). Comparing different serum concentrations (5, 10, 15, and 20%), irrespective of cell sources, the highest proliferation rate was achieved in the presence of 20% serum (p<0.001). Additionally, there was an inverse relation between cell seeding density at culture initiation and proliferation rate, except for L-MSC at 300 cell seeding density.
Conclusion: All three sources of fetal sheep MSCs had the identical trilineage differentiation potential. The proliferative capacity of liver and bone marrow derived MSCs were similar at different cell seeding densities except for the higher fold increase in B-MSCs at 2700 cells/cm2 density. Moreover, the adipose tissue derived MSCs had the lowest proliferative indices.
Introduction :
Mesenchymal Stem cells (MSCs) are more homogenous sub-population of mononuclear cells and comprise a rare population of multipotent progenitors. These cells are capable of both supporting haematopoiesis and differentiating into different tissues originating from mesoderm ranging from bone and cartilage to cardiac muscle 1,2.
The main factors required for a cell to be regarded as a mesenchymal stem cell are their adhesion potential in monolayer culture during in vitro conditions, maintain their undifferentiating characteristics during extended passaging, and differentiation potential into chondrocytes, osteocytes and adipocytes in vitro 3-5.
These adult stem cells are attractive candidates for cell-based therapeutic strategies, substantially because of their easy isolation, purification and amplification, as well as their intrinsic ability to self bio-preserved with minimal loss of potency, their multipotential differentiation, and their amenable to genetic manipulation without adverse reactions to allogeneic versus autologous MSCs transplants 5,6.
MSCs have been identified and isolated from large variety of tissues such as human amniotic fluid 7, placenta 7, Umbilical Cord Blood (UCB) 8,9, veins 10, peripheral blood 11, muscle 12 pancreas, skin, neuronal system 13, dental tissue 14 as well as several fetal tissues including bone marrow 15, blood 15, liver 15, lung, spleen 16, and synovium 17.
In the ensuing decades, extensive research has gone into unlocking the therapeutic potential for MSCs. Bone marrow stroma is the most common sources of these cells for clinical use and designated as the gold standard 7,18. This source of MSCs was first identified in 1960s by Ernest A McCulloch and James E 19. Further experiments in the 1970s and 80s by Friedenstein et al expanded upon the potential of MSCs by demonstrating their capacity for self-renewal and multilineage differentiation 20,21.
Although bone marrow provides a universal source of MSCs, but due to certain shortcomings of obtaining the MSCs including pain, morbidity, low cell number upon harvest, high degree of viral contamination and decrease in the proliferative/ differentiation capacity along with age, alternate sources for MSCs have been sought and subjected to intensive investigation 14,21.
Among different sources of MSCs, adipose tissue, like bone marrow, is derived from the embryonic mesenchyme and possesses abundant and easily accessible MSCs with less invasive method 22 besides its easily growth under standard tissue culture conditions 23.
Recent advances in cosmetic surgery add to its advantage with huge amount of available fatty tissue. Moreover, it has a further advantage when the morbidity associated with large volume bone marrow harvests is taken into the consideration. Its multilineage differentiation has been first identified by Zuk et al 22.
One alternative source is liver tissue. Different types of fetal liver (stem) cells during development were identified, and their advantageous growth potential and bipotential differentiation capacity towards liver or biliary cells has been shown 24-26. Moreover, there are evidences indicating the higher proliferative capacity and immunosuppressive effect of fetal MSCs compared to the adult MSCs 27. In this context there has been an increasing interest in recent years in development of cell based therapeutic strategies as a novel approach in tissue engineering for the treatment of liver diseases 26. In addition, MSCs appear to share similar characteristics across species, which has facilitated the application of MSC in translational studies using animal models. The aim of the present study was to compare growth characteristics and multilinage differentiation potential of sheep foetal MSCs derived from bone marrow (B-MSCs), liver (L-MSCs) and adipose tissues (A-MSCs).
Materials and Methods :
Except where otherwise indicated, all chemicals were obtained from the Sigma (St. Louis, MO, USA). Ovine 30-35 day fetus was obtained from the slaughterhouse and transported to the laboratory in Dulbecco’s PBS (DPBS) with penicillin/streptomycin on the ice.
Bone marrow cell culture: Bone marrow was collected by flushing femurs and tibias with DMEM (Dulbecco’s modified Eagle’s medium) diluted with Phosphate Buffer Solution (PBS 1:2) and supplemented with 100 IU/ml penicillin, 100 IU/ml streptomycin. Mononuclear cells fraction were harvested by Ficoll separation of marrow cells (20 min, 600 g). After separation of cloudy corona and dilution with PBS, it was centrifuged in 200 g for 10 min and washed three times in 4-5 ml PBS. The cells were then incubated in complete medium composed of DMEM, 10% FCS, NEaa , NaHCO3 (3.7 mg/ml), L-glutamin and penicillin/streptomycin at a cell density of 5 x 106 cells/mL in 5% CO2 and 37°C. The adherent elongated cells, MSCs, were exhibited homogeneous fibroblast-like morphology with a spindle or triangular-shaped cell bodies, large and ellipse nuclei and growing outward in a "swirling fibroblast-like" pattern. The nonadherent cells were removed after 48 hr with medium change. After 3-4 days the cultures at 80-90% confluency were tripsinized using 0.05% trypsin/1 mM EDTA and then passaged at 1:2 ratios into fresh 25 cm2 culture flasks. Subculture was repeated till passage 3 when sufficient cells were provided for the next stage of experiment.
Adipose tissue cell culture: The adipose tissue from lumbar paravertebral regions were separated and collected in 15 ml sterile tubes containing PBS supplemented with BSA (20 mg), 100 IU/ml penicillin (Sigma, USA) and 100 IU/ml streptomycin (Sigma, USA). After washing 2 times with PBS, under laminair hood, the specimen was minced into small pieces and separate fibrous tissue.
The specimen subjected to enzymatic digestion using collagenase type IV (0.6 mg) at 37°C for 120 min. At the end of this time, the floating cells were separated from the vascular stromal fraction by centrifugation in 200 g for 10 min. The pellet (stromal vascular fraction) washed 2 times and filtered through a 150 μm nylon mesh to remove undigested tissue. Mononuclear cells were harvested by Ficoll separation to obtain the mononuclear fraction of marrow cells. After separation of cloudy corona and dilution with PBS, centrifuged in 200 g for 10 min and washed tree times in 4-5 ml PBS. The cells were suspended in proliferation medium including DMEM (Dulbecoo Modified Eagle Medium, Sigma, USA) containing 10% FCS (fetal bovine serum, Gibco, Germany), NEaa, NaHCO3 (3.7 mg/ml), L- glutamin and penicillin/ streptomycin (100 U/mL and 100 mg/mL, respectively) (Sigma, USA) and plated at 106 cells/ml in 25 cm2-culture flasks. The cultures were incubated in an atmosphere of 5% CO2 and 37°C. Meanwhile 45-48 hr after culture initiation, the medium was discarded and the cells were washed with PBS and fed with fresh medium. Medium changes were performed each 3 days until the culture became confluent. At this time, the cultures were tripsinized using 0.05% trypsin/1 mM EDTA and passaged at 1:2 ratios into fresh 25 cm2 culture flasks. Subculture was repeated till passage 3 when sufficient cells were provided for the next stage of experiment.
Liver cell culture: The liver from the fetus was dissected with great care without any rush and placed in 15 ml sterile tube containing Hank's balanced salt solution without calcium and magnesium supplemented with 100 IU/ml penicillin and 100 IU/ml streptomycin with pH adjusted to 7.3 and washed 2 times with PBS. Under laminar flow hood, the specimen was mechanically minced into small pieces and then subjected to enzymatic digestion using collagenase IV (0.6 mg/ml) at 37°C for 75 min. At the end of this period, the floating cells were separated by centrifugation at 200 g for 10 min. The pellet was then washed 2 times with PBS and filtered through a 150 μm nylon mesh to remove undigested tissue.
Mononuclear cells fraction were harvested by Ficoll separation (20 min, 600 g). After separation of cloudy corona and dilution with PBS, it was centrifuged in 200 g for 10 min in 4-5 ml PBS (three times). The cells were then suspended in proliferation medium including DMEM containing 10% FCS, NAaa, NaHCO3 (3.7 mg/ml), L-glutamin and penicillin/streptomycin and plated at 106 cells/ml in 25 cm2-culture flasks. The flasks were then incubated in an atmosphere of 5% CO2 and 37°C. After 45-48 hr of culture, the medium was removed and the cells were washed with PBS and refreshed with fresh medium. Medium refreshment was performed after 3 days until the cells became confluent. At this time, the cultures were tripsinized using 0.05% trypsin/1 mM EDTA and passaged at 1:2 ratios into fresh 25 cm2 culture flasks. Subcultures were repeated till passage 3 when sufficient cells were provided for the next stage of the experiment.
Verification of MSCs: In addition to identification of MSCs based on their morphologic or phenotypic characteristics, their multilineage differentiation capacity into the bone, fat, and cartilage were evaluated. Moreover, the stemness property of MSCs and the expression of one related gene to each cell lineage were confirmed by molecular approach.
Osteogenic potential: Osteogenesis of MSCs was induced in vitro by treating a monolayer culture with a pro-osteogenic cocktail. In brief, MSCs in passage 3-4 were cultured at 3×104 cells in 6 well plates containing proliferation medium and allowed to attain 70-80% confluency. The medium was then replaced by differentiating medium composed of DMEM medium supplemented with 10% FCS, 50 μg/ml ascorbic 2-phosphate (Sigma, USA), 10 nM dexamethasone and 10 mM β glycerol phosphate. The cells were kept in differentiating medium for 28 days with medium changes of twice weekly. At the end of this period, the number and size of mineralizing nodules were maximized. For evaluation of mineralized matrix, cells were stained with 2% Alizarin-red S solution. Briefly, the differentiated cells were washed twice with PBS and fixed with 10% formalin for 10 min at room temperature. The cells were then washed thoroughly with PBS and stained with 2% Alizarin Red S solution, pH=4-4.1, with 0.5% NH4OH for 2-5 min. The mineralized matrix was identified by the presence of red foci in stained specimen.
Adipogenic potential: To promote adipogenic differentiation, MSCs at passage 4 were plated in a concentration of 3×104/ml in 6 well culture plates until 70-80% confluency. The proliferation medium was replaced with adipogenic differentiating medium consisted of DMEM supplemented with10% FCS, 50 μg/ml ascorbic 2-phosphate, 100 nM dexamethasone and 50 μg/ml indomethacine. The cultures were kept for 21 days during which the medium was changed twice weekly. The first sign of the adipocyte differentiation became evident 2-3 weeks after culture when lipid droplet appeared in the differentiating cells. Eventually, the lipid-rich vacuoles within cells were combined together and filled the cells. Accumulation of lipid in these vacuoles was assayed histologically by oil red O staining. Briefly, the intracellular accumulation of lipid-rich vacuoles was stained with 0.3% Oil Red O solution. To this end, the cells were fixed in 10% formalin for 10 min, washed with PBS and stained with 0.3% oil red O solution for 10 min at room temperature. The intracellular lipid-rich vacuoles were stained as red foci.
Chondrogenic potential: Chondrogenesis of MSCs in vitro mimics that of cartilage development in vivo. To induce chondrogenic differentiation, the passage 4 MSCs at a concentration of 3×104 cells/ml were plated in 6 well culture plates until 70-80% confluency. The proliferation medium was replaced with chondrogenic medium consisted of high-glucose DMEM supplemented with 0.1 µM dexamethasone, 10 ng/mL TGF-1, 50 μg/ml Ascorbic acid and 50 mg/mL ITS+ premix (Becton Dickinson), 6.25 µg/mL insulin, 6.25 µg/mL transferrin, 6.25 ng/mL selenius acid, 10% FCS at 37°C for 3 weeks. Medium changes were carried out twice weekly and chondrogenesis was assessed at weekly intervals. For the presence of glycosaminoglycans within the extracellular matrix the cells were stained with toluidine blue. Briefly, the differentiated cells were fixed with 10% formalin for 10 min at room temperature. After washing the cells were exposed to Toluidine blue for 30 s at room temperature. Acid mucopolysaccharides and sulfated mucopolysaccharides within the extracellular matrix were stained as violet foci.
Molecular verification of MSCs: The RT-PCR analysis was performed to assess an expression of osteocyte, adipocyte, and chondrocyte related genes in differentiated cell lineages (one gene for each cell lineages) as well as two genes related to the stemness status of MSCs. Total RNA was extracted using RNA Extraction Kit (Rima zol; CinnaGen, Tehran, Iran) according to the manufacturer’s instructions.
Before RT, the extracted RNA samples were treated by RNase-free DNaseI (EN0521; Fermentas, Opelstrasse 9, Germany) to ensure that the extracted RNA for synthesis of cDNA is free of DNA contamination. The extracted RNA was reverse-transcribed to cDNA using 1 mg of extracted RNA, random hexamer primers for ovine genes, and M-MuLV Reverse Transcriptase RNase H- (Vivantis Technologies Sdn. Bhd. Selangor D.E. Malaysia). The PCR reactions were performed using an Eppendorf Mastercycler using primer sequences listed in table 1. The GAPDH was considered as a housekeeping gene. PCR products were analysed in 1% agarose gel, stained with ethidium bromide and visualized by Uvitec gel documentation system.
Growth characteristics: Colonogenic assays (Colony Forming Unit: CFU): To assess the capacity and efficiency for self renewal, passaged-3 MSCs obtained from bone marrow, adipose, and liver tissues were trypsinized and counted using hematocytometere, plated at 100 cells in 10 cm petri dish and allowed to grow for 9 days. At the end of this period, the plates were stained with 1% crystal violet in 100% methanol for visualizing new fibroblast shape colonies (more than 20 cells) produced at cultures. The petri dishes were then observed with a light microscope to determine the number of produced colonies. Calculation of the CFU-F efficiency was performed according to the following formula: CFU-F efficiency=(counted CFU-F/cells originally seeded)×100. Routinely, five CFU-F assays were performed for each isolated cell population.
Population doubling time (PDT): The passage-3 MSCs at a concentration of 104 cells/cm2 were plated in 25 cm2 culture flasks and then cultured until 100% confluency. At this time, the cells were trypsinized and counted with hemocytometer. The PDT was calculated for either studied MSCs according to the following equation: PDT=culture time (CT)/ population doubling number (PDN). To determine PDN, the formulae PDN= log Ni/Nf×3.31 was used 17. In this equation Ni and Nf were the numbers of initiating and harvesting cells, respectively. Routinely, population doubling time assays were performed in triplet for each type of MSCs.
In the following equation; t= culture time, Nh and Ni are the numbers of harvesting and initiating cells, respectively. PDT=(log 2×t)/ (log Nh - log Ni).
Cell seeding density: The MSCs (passage-3) cells were plated at densities of 100, 300, 900, 2700, and 8100 cells/cm2 in 6-well culture plates and cultured in DMEM supplemented with 10% FBS, 100 IU/ml penicillin and 100 mg/ml streptomycin. Five days after culture, the cells were trypsinized, lifted and counted with hemocytometer. Fold increase were then calculated for each cell density.
Serum concentration: MSCs expansion is strongly dependent on FBS presence in medium. To determine optimal serum concentration, passaged 3 cells were plated at density of 16х103 cells/cm2 in 24-well culture plates with the DMEM medium supplemented with FBS concentrations of 5%, 10%, 15% and 20%. When one of the wells in each group reaches confluent (7 day), all cells were trypsinized and counted with hemocytometer. Fold increase in cell number was determined for each culture group and in this regard, the FBS concentrations were statistically compared.
Growth curve: The cultured cells are usually grown with characteristic pattern in which three phases including lag, log and plateau phase can be recognized. In the present investigation, growth curve was plotted for each MSCs derived from bone marrow, adipose tissue, and liver in order to better compare growth kinetics of the cells. For this purpose, the passaged-3 cells derived from each tissue were plated at 3×104 cells/well in 24-well culture plates and allowed to become confluent. In a regular daily basis, some wells were trypsinized and the cell number was determined by hemocytometer count. Using the data growth curves was plotted.
Results :
MSCs growth characteristics: MSCs were successfully isolated and expanded in vitro from all 3 sources. In primary cultures, the cells grew rapidly as such the media change for removal of nonadherent cells was carried out 48 hr of culture and 100% confluency was achieved on day 4 in liver and bone marrow MSCs and day 5 of the culture in A-MSCs (Figure 1). The MSCs were kept their phenotypically homogeneous morphologies as well as their multiplication characteristics in cultures in different sequential passages.
Assessment of PDT in both MSCs lines was revealed no difference in PDT between two sources of MSCs when the cells plated at 104 cells/cm2 in a culture medium supplemented with 10% FBS (Table 2).
Concerning the colonogenic capacity of MSCs, no difference was observed in the number of fibroblast shape colonies (each more than 20 cells) following 100 cells seeding in 10 cm petri dish after 9 days between bone marrow and liver derived MSCs (Table 2, Figure 2).
In MSCs lines, (liver and adipose tissue), the maximum proliferation of the cells (fold increase) was achieved when cell seeding was adjusted to 300 cells/cm2 in the presence of 10% FBS (p<0.001). Indeed, as the cell seeding density was increased the fold increase was decreased except for the 300 cells/cm2 (Table 3). There was no significant difference in fold increase in different cell seeding densities between bone marrow and liver derived MSCs except for the 2700 cells/cm2, in which the fold increase in bone marrow derived MSCs was higher than liver MSCs (p<0.01).
The cells having been plated with 20% FBS had a significantly larger fold increase in cell number as compared with lower concentrations of serum (Table 4). According to the plotted curve, the cells in different sources started proliferating immediately after being plated. The culture reached plateau in approximately 5-9 days after initiation depending on the cell sources (Figure 3).
Multilineage cell differentiation: In osteogenic media, the morphology of MSCs derived from three sources was changed from spindle-shaped to cubical cells and the mineralized foci were detected as red spotted areas stained with Alizarin red (Figure 4). The induced chondrogenic differentiation was detected by the presence of glycosaminoglycans within the extracellular matrix stained with toluidine blue as violet foci (Figure 5). The differentiated MSCs to lipocytes, underwent morphological changes and produced vacuoles containing lipid droplets detected with oil red O staining as red areas (Figure 6).
MSCs molecular verification: At the molecular basis, RT-PCR, the osteogenic, lipogenic, and chondrogenic differentiation potential of MSCs derived from different sources was further confirmed by expression of specific genes related to differentiated cell lineages (one gene for each cell lineage). The stemness status of isolated MSCs, from 3 sources, was confirmed by expression of 2 related genes (Figure 7).
Discussion :
Physiological properties of mesenchymal stem cells including straightforward manipulation, high proliferative capacity, immunomodulation, multilineage differentiation potential, and tropism for the healing of injuries render them a potentially powerful candidate cell type for regenerative medicine as well as for the study of cellular differentiation 5,28. The plasticity, self-renewal, and multi-lineage potential of MSCs have generated growing interest in their use in a constantly expanding variety of experimental regenerative therapies and transplantation purposes 29,30. Nevertheless, further studies in suitable animal models are needed to translate the potential of MSCs into clinical applications.
The discovery of mesenchymal stem cells is credited with Alexander Friedenstein and associates, who over 46 years ago demonstrated that pieces of bone marrow transplanted under the renal capsule of mice, formed a heterotopic osseous tissue that was self-maintaining, self-renewing, and capable of supporting host cell hematopoiesis 20. Furthermore, Friedenstein showed that the osseous-forming activity of bone marrow was contained within the fibroblastoid cell fraction isolated by preferential attachment to tissue culture plastic. Nowadays, B-MSC has been the traditional source of human MSCs (hMSCs) for basic research and therapeutic purposes because of its routine harvest and safe procedure 20,31.
According to the previous investigations, MSCs are present at low frequencies in bone marrow samples 1,32. Therefore, finding alternative sources of mesenchymal stem cells for research and therapeutic purposes are necessary. In the present study, adherent spindle-shaped cells derived from fetal ovine liver, adipose tissue and bone marrow were collected and examined in terms of their growth characteristics, culture requirements and tripotent differentiation potential that is proposed as a criterion indicating MSCs identity of the cells in question.
These cells show a varying cellular morphology from spindle-shaped and elongated, that mostly were observed, towards more cuboidal fibroblast-like cells with shorter cytoplasmatic extensions (Figure 1). More specifically, Sekiya and colleagues demonstrated that hMSCs undergo a time-dependent morphological transition from thin (small), spindle-shaped cells (considered stem cells or early progenitors) to wider (larger) spindle-shaped cells (looked like more mature cells) when cells were plated at 1 to 1000 cells/cm2 33. Moreover, Jung et al showed that the small, spindle-shaped cells proliferate more rapidly and have a higher level of multipotentiality, compared to the slowly replicating large cells, which have lost most of their multipotentiality 34.
The morphology and size of hMSCs may also be dependent upon culture conditions (e.g., growth media, culture surface). For instance, the cultured hMSCs in bFGF-supplemented media were smaller and proliferated more rapidly than those cultured in bFGF-lacking conditions 35. Culture surfaces (e.g., treated with Matrigel) might also affect the morphology 36.
The ability of cells to reform colonies, a primitive measure of progenitor cell activity, was initially determined by using a colony-forming assay. Considering the growth characteristics of three sources of MSCs the highest PDT and lowest colony forming unit were observed in A-MSCs. Therefore, it could be inferred that whenever the fast propagation of MSCs is demanded adipose tissue is not a good candidate compared to the liver and bone marrow. Though, the ease of access and the sufficient amount of tissue in isolation and purification of MSCs is another factor that should be considered in choosing the source of MSCs (Table 1).
The colony numbers and PDT in B-MSCs and L-MSCs were comparable to the corresponding values in MSCs derived from different sources in other species 37-40. Morphologically, human and porcine bone marrow MSCs comprised of approximately 2-3% of the total nuclear cell fraction in the bone marrow with PDT of 30-50 hr and 50-55 hr, respectively 38,41,42. The B-MSC PDT in rat is varied between 24 hr to 62 hr 39,43. In pubertal sheep, the B-MSCs PDT was 24.94 hr when plated at 100 cells/cm2 32, though in another report it was at about 50 hr 4 which was slightly different from the results of our study (31 hr).
The PDT in fetal L-MSCs was slightly lower than what reported in another study (28 hr vs. 36 hr) 44. It seems apart from the tissue source of MSCs, the site from which the tissue is obtained can influence the PDT. In this context, the PDT in epididymal and epicardial adipose tissue derived stem cells has been reported 69±16 hr and 45±9.6 hr, respectively 45.
There are several approaches to improve conventional tissue culture conditions in order to get a higher number of cells and to prevent loss of their differentiation capacity. Studies on the effect of serum source 46,47 and the use of growth and differentiation factors like, bone morphogenetic proteins and fibroblast growth factors are all of high concerns 48,49.
FBS contains a high content of growth factors as well as nutritional and physiochemical compounds required for cell maintenance and growth 34. Therefore, cell propagation is largely dependent on the presence of FBS in culture medium 1,40. FBS-based media is a common standard medium used for propagation of hMSCs for basic research and clinical studies 34,46. Therefore, any experimental work with MSCs requires in vitro expansion of the cells with the most appropriate concentration of serum.
According to the previous investigations, the number and proportion of MSCs in different tissues is quiet low, therefore, their expansion is necessary before performing clinical studies 32. To do so, we cultivated three sources of ovine MSCs in the presence of different concentrations of FBS to determine the least appropriate serum concentration with the highest mitogenic effect. This is of utmost importance especially at cell therapy strategies where the rapid expansion of cells would be desired. In our study condition, there was a positive relationship between serum concentration and proliferative capacity of MSCs. The MSCs exhibited the highest proliferation when being provided with a medium supplemented with 20% FBS. This finding was in contrast to other study in which the maximum fold increase in ovine and goat MSCs was achieved at 15% serum concentration 32,40. There was, however, no significant difference between 15% and 20% serum in rat and mouse MSCs propagation 50,51.
Concerning the effect of serum on cell propagation one should bear in mind how serum concentration may influence the quality, functionality, immunomodulatory properties, and differentiation potential of MSCs in in vitro and in vivo conditions, especially in tissue engineering.
The other factor affecting the rate of MSCs proliferation would be the cell seeding density at culture initiation. The cell proliferation rate is of utmost importance especially at cell therapy strategies where the rapid expansion of cells would be desired and can be used to optimize the culture condition for maximum proliferation of the cells 45. Based on our findings, there was an inverse correlation between MSCs seeding density and their proliferation which was manifested as a reduction of fold increase concurrent with the increasing of cell seeding density at culture initiation, except for L-MSC at 300 cell seeding density (Table 3). Moreover, in B-MSCs the fold increase at 300 cells/cm2 cell density, though insignificant, was higher than 100 cells/cm2 cell density. This finding, somehow, was in agreement with the reports indicating 100 cells/cm2 cell density is superior in B-MSCs proliferation in sheep 32, goat 40, mouse 51, and rat 50 species.
Despite the higher fold increase at lower densities and considering the finite lifespan of MSCs, the question of how the higher population doubling in lower densities groups may influence the quality and differentiation potential of MSCs remains to be investigated. On the other hand, whether the proliferated cells at lower densities, at culture initiation, have proliferation and differentiation potential similar to cells with higher densities is controversial.
Concerning the multilineage differentiation potential of the three sources of MSCs, their multilineage differentiation characteristics were confirmed by their differentiation into osteoblasts, adipocytes, and chondrocytes under appropriate culture conditions. Additionally, the differentiation status of MSCs was further confirmed by mRNA expression analysis. No difference was observed in multilineage differentiation potential and mRNA expression analysis between different sources of MSCs.
Conclusion :
In conclusion, in our study condition all the three sources of fetal sheep MSCs had the same multilineage differentiation potential. Among the three sources of MSCs the bone marrow and adipose tissue derived MSCs had the highest and the lowest proliferative potential, respectively. In all the three sources of MSCs, there was a negative relationship between cell seeding density at culture initiation and proliferative capacity except for the liver and bone marrow derived MSCs at 300 cells/cm2 density. Considering both the PDT and proliferative potential where the rapid expansion of cells would be desired, the liver derived MSCs are good alternative for bone marrow derived MSCs.
Acknowledgement :
The authors would like to thank the Avicenna Research Institute for technical and financial supports, ACECR, Tehran, Iran.

Figure 1. Primary culture of sheep mesenchymal stem cells. At primary culture (48 hr after culture initiation) adherent spindle-shaped cells showed a varying cellular morphology from spindle-shaped (arrow) towards more cuboidal fibroblast-like cells (arrowhead) in adipose tissue, A) (400×), liver; C) (200×), and bone marrow; E) (200×) derived MSCs. Elongated fusiform cells were mostly observed after day 5 of the culture in adipose tissue; B) and after day 4 in liver; D) and bone marrow; F) derived MSCs (200×)
|
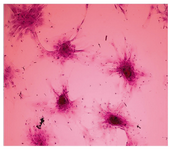
Figure 2. Efficiency of self renewal of MSCc assessed by rate of colony formation in CFU-F assays. Crystal violet staining for examination of CFU after 21 days showed no difference in the number of fibroblast shape colonies (each more than 20 cells) between bone marrow and liver derived MSCs (100×)
|
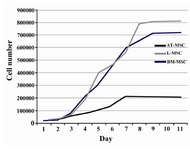
Figure 3. Growth curve of MSCs. The cells of different sources started proliferation immediately after being plated and reached plateau in approximately 5-9 days after culture initiation depending on the cell sources
|
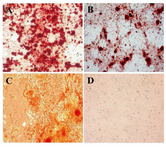
Figure 4. Osteogenic differentiation after 28 days showed marked morphological changes and extensive extracellular calcium deposition in bone marrow; A) Liver; B) and adipose tissue; C) derived MSCs as demonstrated by positive Alizarin Red S staining (200×). No mineralized nodules were apparent in non induced cell cultures; D) (200×)
|
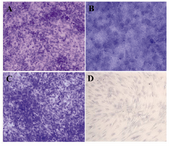
Figure 5 Chondrogenic differentiation after 21 day. In chon-droinductive culture, the metachromatic nature of the matrix was determined by the toluidine blue staining of glycos-aminoglycans as purple stained loci in bone marrow; A) (200×) liver; B) (400×) and adipose tissue; C) (200×) derived MSCs. No stained foci were observed in non induced cell cultures; D) (400×)
|
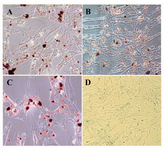
Figure 6. Adipogenic differentiation after 21 days. In adipogenic cultures, formation of lipid droplets within the cytoplasm of the cells was quite slow and were gradually occupied the whole cells. Accumulation of intracellular lipid droplets was determined as red loci following oil red O staining in bone marrow, A) Liver; B) and adipose tissue; C) derived MSCs (400×). No stained foci were observed in non induced cell cultures; D) (200×)
|
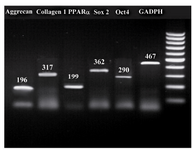
Figure 7. Representative gel photographs of RT-PCR analysis of important gene transcripts related to stemness status of MSCs and one related gene to each cell lineage (adipocyte, osteocyte, and chondrocyte). GAPDH was considered as re-ference gene
|

Table 1. Details of primers used for RT-RCR
|
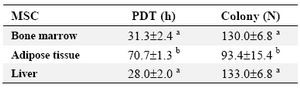
Table 2. Colony numbers and population doubling time in mesenchymal stem cells derived from different sources
a, b) numbers with different superscript in the same column differ significantly p<0.001
|
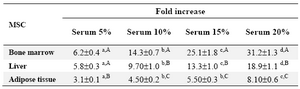
Table 3. The effect of serum concentration on proliferation of mescnchymal stem cells derived from different sources
a-d) Numbers with different lowercase superscript letters in the same row differ significantly (p<0.001)
A-C) Numbers with different uppercase superscript letters in the same column differ significantly (p<0.001)
|
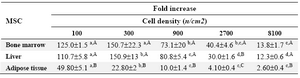
Table 4. The effect of cell seeding density on proliferation of mesenchymal stem cells derived from different sources
a, d) numbers with different lowercase superscript letters in the same row differ significantly (p<0.001)
A,C) numbers with different uppercase superscript letters in the same column differ significantly (p<0.01)
|
|