Construction and Evaluation of an Expression Vector Containing Mtb32C (Rv0125) of Mycobacterium tuberculosis
-
Nabavinia, Maryam Sadat
-
Microbiology and Virology Research Center, Avicenna Research Institute & Department of Medical Bacteriology & Virology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
-
 Naderi Nasab, Mahboobeh
Microbiology and Virology Research Center, Avicenna Research Institute, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran, Tel: +98 511 8022206 Fax: +98 511 7636185 E-mail: naderinasabm@mums.ac.ir
Naderi Nasab, Mahboobeh
Microbiology and Virology Research Center, Avicenna Research Institute, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran, Tel: +98 511 8022206 Fax: +98 511 7636185 E-mail: naderinasabm@mums.ac.ir
-
Microbiology and Virology Research Center, Avicenna Research Institute & Department of Medical Bacteriology & Virology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
-
Meshkat, Zahra
-
Microbiology and Virology Research Center, Avicenna Research Institute & Department of Medical Bacteriology & Virology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
-
Women's Health Research Center, Mashhad University of Medical Sciences, Mashhad, Iran
-
Derakhshan, Mohammad
-
Microbiology and Virology Research Center, Avicenna Research Institute & Department of Medical Bacteriology & Virology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
Abstract: Expressions of recombinant proteins for different applications are important objectives in molecular biotechnology; however, expression of some recombinant proteins is difficult. Several methods have been designed for expression of these proteins. The aim of this study was to construct a vector containing Mtb32C fragment of Mycobacterium tuberculosis (M.tuberculosis) as a fusion partner in order to improve the expression of fused recombinant proteins. Mtb32C was amplified by polymerase chain reaction (PCR). The amplified fragment was ligated into pET21b+ vector. Colony-PCR, enzyme digestion and DNA sequencing methods were used to confirm the recombinant vector. Colony-PCR showed a 420 bp fragment in size corresponding to the correct size of our fragment. In addition the recombinant plasmids sequencing showed the accuracy of the cloned fragment. For confirming the expression, reverse transcriptase (RT)-PCR analysis was performed showing a 420 bp fragment in agarose gel electrophoresis using specific primers. The construction of a vector containing Mtb32C fragment is promising as a fusion partner for future studies as it affected the expression of the fused proteins and increased immune responses against the partner.
Introduction :
Recent progresses in biotechnology sciences have resulted in development of recombinant proteins. Also their applications in biochemical and structural studies of the proteins and treatment purposes have increased. The first step in producing the recombinant proteins is the cloning of the desired gene into a suitable vector for expression of the protein. This is then followed by other steps such as purification, identification and structural and/or functional analysis (1). In addition, solubility of the proteins may be considered as an important property (2). However, high and rapid expressions of the proteins as well as the purification steps are major problems in the study of recombinant proteins (3).
Escherichia coli (E.coli) is a gram-negative bacterium that is used widely as a host for prokaryotic expression of the recombinant proteins in both industrial and laboratory scales. There are several advantages for application of E.coli as a host for production of the recombinant proteins; E.coli was the first organism that its genome was recognized and sequenced. Therefore, several methods have been developed to improve the gene cloning and protein expression in the E.coli. In addition, E.coli grows rapidly at high density and it needs minimal and inexpensive substances for propagation (4).
Application of fusion partner is a successful method for increasing the protein production (5,6). Several fusion partners have been designed to facilitate the expression of the recombinant proteins. Previous studies showed the Mtb32C of Mycobacterium tuberculosis (M.tuberculosis) when used as a fusion protein at the N terminal of recombinant proteins could increase their production and also their immune responses against the fused antigen compared to the antigen alone without Mtb32C tag (7).
The aim of this study was cloning of Mtb32C into pET21b vector and evaluation of its ability to express the cloned fragment, Mtb32C, in order to construct a suitable vector which can be used for increasing the expression of desired recombinant proteins in future. In addition, the designed vector can be used for enhancing the immune responses against its fusion partners.
Materials and Methods :
Mtb32C gene was amplified by Polymerase Chain Reaction (PCR) method from genomic DNA of M.tuberculosis H37Rv genome (Pasture Institute of Iran). Two specific primers, 5´ CAATTACATATGACGGCCGC GTCCGATAACTTC3´ as a forward primer and 5´ CTAATCGGATCCGGCCGGGGGTC CCTCGGCCAA3´ as a reverse primer, were used for PCR amplification (underlined letters in forward and reverse primers show NdeI and BamHI restriction enzyme sites, respectively). The PCR was performed as described previously (8). About 150 µl of PCR product was used for agarose gel electrophoresis and purified by Bioneer extraction kit (Korea). The extracted fragment was digested using NdeI and BamHI restriction enzymes (Roche) according to the manufacturer's instructions.
The next step was insertion of the digested and purified fragment (Mtb32C) into the digested and purified vector (pET21b).
Competent bacteria strain DH5α were prepared by cold CaCl2 0.5 M and transformed by heat shock method (90 s at 42°C) (1). Transformed bacteria were cultured on LB agar containing 100 µg/ml ampicillin and incubated at 37°C about 16 hr. The presence of the Mtb32C into the recombinant vector was confirmed by colony-PCR using specific primers, restriction enzyme analysis and sequencing (Milligene Company, France).
For confirming the protein expression, BL21b strain of the E.coli was cultured in 2*YT medium and the competent cells were prepared using CaCl2 as described before. The Mtb32C vector was transformed into competent E. coli BL21b by heat shock method. The transformed bacteria were cultured in 2*YT medium containing 50 µg/ml ampicillin. Isopropyl-β-D-thio-galactoside (IPTG) 2 µM was used for induction of the bacteria (OD 0.5 at 600 nm) for protein expression. Four hr after induction, the bacteria were washed with PBS and their RNA was purified using RNX Plus kit (Cinagen Company, Iran) according to the manufacturer's instructions. The extracted RNA was used in RT-PCR as described previously (9). In brief, 1 µg total RNA was used for cDNA synthesis and PCR was performed on cDNA by the Mtb32C specific primers as described above.
Results :
PCR method was used for amplification of Mtb32C fragment and the amplified product was analyzed by electrophoresis on 2% agarose gel. The PCR result showed a 420 bp band on the gel (Figure 1).
The Mtb32C fragment was prepared in large scale by PCR method and after purification it was inserted into the pET21b cloning vector. Then E.coli DH5α competent bacteria were transformed by the recombinant cloning vector. After 16 hr incubation at 37⁰C, six colonies were grown on LB agar containing ampicillin.
The presence of desired plasmid was confirmed by colony-PCR using Mtb32C specific primers. The colonies containing desired plasmid were positive in colony-PCR and their fragment was 420 bp (Figure 2).
The positive colonies were selected for further analysis. At first, the colonies were propagated and purified then used for restriction enzyme digestion and sequencing. Restriction enzyme digestion results showed the molecular weight of pET21b vector containing the Mtb32C was more than the vector without the fragment. To check the protein expression, RNA was extracted from the E.coli BL21b strain and RT-PCR was performed using the Mtb32C specific primers which showed the expected size in agarose gel electrophoresis (Figure 3).
DNA sequencing results were compared to the gene sequences that were present in the NCBI Nucleotide database and the result of the NCBI BLAST showed that mutation (insertion, deletion, substitution, …) did not occur during gene cloning. In addition, the sequence showed 100% similarity with the Rv0125 (Mtb32) gene of M.tuberculosis H37Rv which was used in this study.
Discussion :
Recombinant protein production and their application in biotechnology have been increased. However, high expression of recombinant proteins in prokaryotic and eukaryotic expression systems is an issue. Different methods including fusion partners have been developed to overcome this limitation. Several fusion partners were designed to increase and facilitate the expression of the recombinant proteins. Bao et al showed B1 immunoglobulin binding domain of Streptococcal protein G (GB1) increased the expression of a recombinant protein in E.coli (10). Panavas et al reported Small Ubiquitin-like Modifier (SUMO) protein enhanced protein production when it was used at the N terminal of the recombinant protein (11). EspA is a secreted protein of the enterohaemorrhagic E.coli. Cheng et al demonstrated the EspA enhanced production, stability and solubility of the foreign proteins in E.coli (12). Furthermore, Maltose-Binding Protein (MBP), transcription termination anti-termination factor (NusA), E.coli thioredoxin (TrxA), and protein disulfide isomerase I (DsbA) were frequently used as fusion partners (13). Previously, Wang et al showed Mtb32C fused to N terminal of an antigen can not only increase levels of protein expression in E.coli, but also can increase host immune responses against the fused antigen (7). In addition, other studies showed administration of a recombinant protein consist out of a Mtb39 gene of M.tuberculosis fused to Mtb32, enhanced immune responses compared with Mtb39 protein alone (8,14). Therefore, cloning of different antigens specially antigens with low immunogenicity in association with Mtb32 can improve the host immune responses against the fused antigen.
This may in future be important in immunological studies especially in recombinant vaccine designing. Mtb32 is a serine protease protein of M.tuberculosis which can stimulate immune responses against the bacterium (8,14). In this study, Mtb32C fragment, C-terminal part of Rv0125 gene of M.tuberculosis H37Rv was amplified and cloned into pET21b(+) vector successfully. In our study, application of NdeI and BamHI restriction enzymes for cloning the Mtb32C fragment leads to remaining of several restriction enzyme sites which can be used for cloning the other genes.
Conclusion :
In summary, the cloning and expression vector containing Mtb32C fragment was constructed in present study. The prepared vector is suitable for application in designing the fusion proteins and recombinant vaccines because it can enhance the expression of fused protein and improve the immune responses against the protein.
Acknowledgement :
This study is part of a thesis presented for the degree of Master of Science (MSc) in Medical Microbiology which was supported by Mashhad University of Medical Sciences, Mashhad, Iran (grant No. 88638 and thesis No. 294-A). We would like to thank Dr Mohammad Reza Razavi, from the Pasture Institute of Iran for providing M.tuberculosis H37Rv genome.
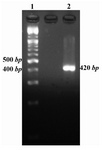
Figure 1. Mtb32C PCR result. Lane 1 is 100 bp DNA size marker and lane 2 is Mtb32C PCR product (420 bp fragment)
|
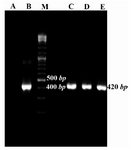
Figure 2. Colony-PCR results for growing colonies using Mtb32C specific primers. Lane A: negative control of PCR (PCR mixture was prepared using water instead of DNA template), B: positive control (purified PCR product from Agarose gel), C, D, and E: positive colonies containing recombinant vector consist of Mtb32C fragment (420 bp fragment), M: 100 bp DNA size marker
|
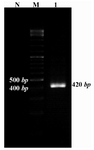
Figure 3. Gel electrophoresis of RT-PCR products. RT-PCR was performed for amplifying 420 bp fragment using Mtb32C specific primers. Lane 1 correspond to the RT-PCR result; M= 100 bp DNA size marker; N= non transformed bacteria as a negative control of expression
|
|