Cloning and Expression of Soluble Recombinant HIV-1 CRF35 Protease-HP Thioredoxin Fusion Protein
-
Azarnezhad, Asaad
-
Cellular and Molecular Research Center, Kurdistan University of Medical Sciences, Sanandaj, Iran
-
Department of Medical Genetics, Faculty of Medicine, Tehran University of Medical Science, Tehran, Iran
-
Sharifi, Zohreh
-
Blood Transfusion Research Center, Institute for Research and Education in Transfusion Medicine, Tehran, Iran
-
Seyedabadi, Rahmatollah
-
Department of Molecular Medicine and Genetics, Faculty of Medicine, Hamedan University of Medical Sciences, Hamedan, Iran
-
 Hosseini, Arshad
Department of Medical Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran, Tel: +98 9128454881, E-mail: ash-hosseini@sina.tums.ac.ir
Hosseini, Arshad
Department of Medical Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran, Tel: +98 9128454881, E-mail: ash-hosseini@sina.tums.ac.ir
-
Department of Medical Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran
-
Johari, Behrooz
-
Department of Medical Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran
-
Sobhani Fard, Mahsa
-
Department of Immunology, Faculty of Medicine, Hamedan University of Medical Sciences, Hamedan, Iran
-
Student Research Center, Hamedan University of Medical Sciences, Hamedan, Iran
Abstract: Background: As a drug target and an antigenic agent, HIV-1 protease (HIV-1 PR) is at the center of attention for designing anti-AIDS inhibitors and diagnostic tests. In previous studies, the production of the recombinant protease has been faced with several difficulties; therefore, the aims of this study were the easy production, purification of the soluble form of protease in E. coli and investigation of its immunoreactivity.
Methods: Protease coding region was isolated from the serum of an infected individual, amplified by RT-PCR and cloned into PTZ57R using TA-cloning. Protease coding frame was isolated by PCR and cloned in pET102/D. TOPO expression vector and cloned protease was expressed in Escherichia coli (E. coli) BL21. Produced recombinant protein was purified by affinity Ni-NTA column and protein concentration was checked by BCA protein assay kit. Subsequently, immunoreactivity of recombinant protease (rPR) was assayed by Western blotting and ELISA.
Results: Cloning of the HIV protease by TOPO cloning system in pET102/D.TOPO was confirmed with PCR and sequencing. The concentration range of purified recombinant protein was 85 to 100 μg/ml. Immunogenicity of rPR was confirmed by Western blotting and ELISA.
Conclusion: Soluble production of recombinant HIV-1 protease (HIV-1 rPR) was performed successfully. This recombinant protein disclosed 86% specificity and 90% sensitivity in immunoassay tests.
Introduction :
Two pathogenic types of Human Immunodeficiency Virus (HIV) are known as HIV-1 and HIV-2. HIV-1 is the predominant virus in most parts of the world 1. At present, HIV-1 phylogenetic classifications are based on genome sequence analysis that leads to identifying of inter-subtype recombinant forms named Circulatory Recombinant Forms (CRFs) 2. CRF35_AD is one of the highly prevalent forms of HIV-1 in Iran and Afghanistan that should be more investigated 3. Following its recognition in 1981, the HIV/AIDS epidemic has evolved to become the greatest challenge in global health, with some 34 million persons living with HIV worldwide 4. HIV diagnosis is very critical for effective preventive strategies and designing a plan for positive cases. HIV/AIDS testing is based on immunoblotting assays (ELISA, Western blotting) and viral nucleic acid test. Also, antiviral therapy in AIDS is based on inhibitors that target the vital component of the virus 5.
HIV-1 is a RNA virus of retrovirus family whose genes are initially transcribed as GAG, POL and ENV poly-protein precursor that needs to be cleaved for completing viral life cycle 6. HIV-1 protease (HIV-1 PR) derived from pol polyprotein is an aspartic protease which acts in homo-dimer form 7-9. It processes the viral gag, gag-pol and env to the mature structural and enzymatically active proteins and it is essential for the viral life cycle, so that point mutations inactivating this enzyme have given rise to the production of noninfectious progeny virus 10. Accordingly, the HIV-1 protease is one of the main targets for anti-HIV drug therapy 11. Although standard treatment with current protease inhibitors is somehow effective, these inhibitors have some limitation such as drug-resistance and non-response 12. Therefore, easy production and purification of large quantities of this enzyme are prerequisites for the development of assays which allow the identification of potent and selective inhibitors.
On the other hand, previous studies have shown that HIV-1 PR can trigger host immune system. This enzyme has several conserved epitopes that play a role in epitope selection and immunodominance 13. Enzyme-linked Immunoassay (EIA) of a bacterially expressed protease indicated that most individuals developed an antibody response to protease. So, protease seems to be quite antigenic 14. Given the importance of AIDS diagnosis for preventing infection transmission and management of infected individuals, heterogeneous production of protease can promote the specificity and sensitivity of immunoblotting and ELISA diagnostic assays.
Because of cytotoxicity and hydrophobicity (low solubility) of protease, it is difficult to obtain large quantities, so that in most cases, the expression level has been low and it can only be detected by immunoblotting 15. Therefore, the main purposes of this study were to construct a high and reproducible expression system for production of recombinant HIV-1 protease (HIV-1 rPR), establish a convenient (simple and fast) purification procedure and finally test the immunogenicity feature of HIV-1rPR using immunoblotting and Enzyme-Linked Immunosorbent Assay (ELISA).
Materials and Methods :
Sample preparation: Serum samples of HIV-1 positive patients were provided by virology lab of the Iranian Blood Transfusion Organization (IBTO). All samples had been confirmed to be either HIV positive or negative by Western blotting and real-time PCR.
Viral RNA isolation and RT-PCR: Viral RNA was isolated from HIV positive serum samples using the Viral RNA extraction Mini Kit (Qiagen, USA) according to the manufacturer’s instructions. Synthesis of cDNA was done under following conditions: 5 μg of isolated viral RNA was added to RT reaction mixture containing 20 units of PrimeScript™RTase (200 U/µl), 100 pmol of random hexamer, 4 μl of 5 x RT buffer, 20 units of Thermo Scientific RiboLock RNase inhibitor, and 10 mM of dNTPs mix (all from Fermentas, Lithuania) adjusted to the final volume of 20 μl with d.H2O. The mixture was then incubated for 10 min at 25°C followed by 60 min at 37°C. Termination of the reaction was done by heating at 75°C for 8 min.
Nested PCR was carried out using cDNA samples with external and internal primer pairs listed in table 1. RT-PCR reaction was performed for achievement of 504 bp A-tailed PCR product using Taq DNA polymerase (Fermentas, Lithuania) according to following thermal cycle program: denaturation at 94°C for 5 min, 34 cycles of 94°C for 1 min, 58°C for 30 s, and 72°C for 60 s. A final extension cycle was run at 72°C for 10 min. The PCR products were purified using PCR Cleanup Kit (Roche, Germany). Then, purified PCR products were cloned into PTZ57R (T) vector using InsTAclone PCR Cloning Kit (Fermentas, Lithuania). All procedures were accomplished according to the instruction manuals provided by kit manufacturers. Confirmation of insertion of the PR coding sequence was carried out by sequencing of PTZ57R-PR and colony-PCR (FAZA Biotech Co, Tehran, Iran).
Cloning and expression of PR coding region in pET102D/TOPO expression vector: The primers set of Fprot and Rprot (Table 1) were designed for amplification of PR coding sequence from PTZ57R-PR. CACC base at the start of forward primer is for direct insertion of blunt end PCR product into TOPO cloning vector. DNA encoding PR was amplified using pfu DNA polymerase (Stratagen, USA) in a gradient master cycler (Eppendorf, Hamburg, Germany) as follows: initial denaturation at 96°C for 4 min, 30 cycles consisted of denaturation at 94°C for 45 s, annealing at 58°C for 30 s and extension at 72°C for 40 s. A final extension cycle was performed at 72°C for 5 min. Amplified PR coding region was purified and inserted into the pET102D/TOPO expression vector (Invitrogen, USA) according to the manufacture instruction, and transformed into BL21 (DE3) Escherichia coli (E. coli) strain. Obtained transformed E. coli was cultured on LB agar medium including 100 ng/μl ampicillin, 40 µl of X-Gal stock solution (20 mg/ml) and 40 µl of IPTG 100 mM. White colonies are considered as recombinant bacteria and both colony-PCR and sequencing were accomplished for analysis of recombinant clones.
True recombinant E. coli BL21 (DE3) was then cultured overnight at 37°C in LB medium containing 100 ng/μl ampicillin. In the following day, 500 μl of overnight culture was added to 250 ml of fresh LB medium in a shaker incubator (150 rpm) at 37°C for 5 hr until the logarithmic phase (OD600=0.5-0.6). E. coli BL21 (DE3) cells were then induced by IPTG at a final concentration of 0.1 M for 4 hr at 37°C. After 4 hr, E. coli pellet was harvested using centrifugation at 3500 rpm (Eppendorf 5804) for 12 min and used for subsequent analysis including expression confirmation, purification and immunoblotting assays.
Expression analysis and purification of recombinant protein: Achieved induced E. coli pellet was sonicated by ultrasonic system UP100H (Hielscher Inc, USA). For this purpose, cell pellet was resuspended in chilled lysis buffer (50 mM Tris-HCl pH=7.5, 100 mM NaCl, 5 mM DTT, 1 mM PMSF) and cooled on ice for 10 min. Then, cell suspension was sonicated with 10 short bursts of 10 s followed by interval of 30 s for cooling. Finally, cell debris was removed by ultracentrifugation at 4°C for 15 min at 14000 rpm. For confirmation of rPR expression, the supernatant was run on 12% polyacrylamide gel and analyzed by SDS-PAGE and Western blotting. Purification of rPR was done using Ni2+-NTA resin (Invitrogen, USA) according to the manufacturer’s guide. In this stage, native purification method was used. The purity of the purified protein was assessed using electrophoresis on the 12% polyacrylamide gel and subsequent Coomassie blue staining. Purified protein concentration was measured by Micro BCA protein assay kit (PIERCE, USA).
Immunoblotting and ELISA: Following the acrylamide gel electrophoresis, the protein bands were transferred to the nitrocellulose membrane. Blocking of the membrane was developed with 5% of skim milk in TBST buffer (Tris-Buffered Saline containing 0.1% v/v Tween 20) for 1.5 hr and incubated overnight in a shaker at 4°C. On the following day, membrane was incubated with human serum of patient infected with HIV-1 diluted 1:12000 in 3% skim milk and TBS buffer for 3 hr on a shaker at 37°C. The membrane was washed 3 times with TBST buffer and incubated with mouse anti human-HRP antibody diluted 1:90,000 in 3% skim milk in TBS buffer for 3 hr at Room Temperature (RT) on a shaker. Again, membrane was washed 3 times with TBST and 1 time with PBS buffer. Thermo Scientific™ Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Wyman Street, Waltham, USA) was added to membrane and blotting results were visualized using a light-sensitive film. In parallel, another Western blot analysis was performed using anti-His tag monoclonal antibody (Invitrogen, Frankfurt, Germany) diluted 1:2000 in 3% skim milk in TBS buffer. This antibody specifically binds to poly-histidine tag of the recombinant protein and confirms the expression of the recombinant protease.
In this study, ELISA was performed to test specificity and sensitivity property of rPR. To do this, MaxiSorp 96-well plates ((Nunc Technologies, Roskilde) were used in which hydrophilic HP-thioredoxin rPR coats its surface with high efficacy. 80 μg.μL-1 purified rPR suspended in 0.1 M PBS (pH=7.5) was used for coating and incubated overnight at 4°C. On the following day, 96 wells of microtiter plate were washed 3 times with Phosphate Buffer Saline (PBS) and blocked with 5% BSA in PBS for 1.5 hr at RT. Plate was washed 5 times with PBST buffer (containing PBS and 0.2 Tween 20). HIV- infected and healthy control sera diluted 1:300 in PBS containing 0.1% (v/v) Tween-20 and 0.01% (w/v) BSA were applied to each well and incubated at 37°C for 40 min. All wells were consequently washed 5 times with PBST, 1:2000 dilution of secondary mouse antihuman-ALP conjugated antibody in PBS added to each well and the plate was incubated for 1 hr at RT. Again, each well was washed 3 times to remove the unbinding antibodies. Finally, the plate was incubated with 100 µl of PNPP for 30 min and applied to ELISA reader (Biochrome, Cambridge, UK) at OD450. Specificity and sensitivity values of rPR were evaluated by 50 positive and 50 negative serum samples. ELISA results were analyzed using receiver operating characteristic (ROC) analysis by SPSS software, version 16.0 (SPSS Inc, Chicago, IL) and cut-off (CO) values for the test were set according to Youden's index. According to this index, the maximum CO value set to 0.78 was equal to the point where Y=sensitivity+(specificity-1).
Results :
RT-PCR and amplification of protease gene from PTZ57R-PR: RT-nested PCR product of the region containing protease gene was visualized on 1% agarose gel (Figure 1A). For this purpose, a fragment with 504 bp was successfully amplified and recombinant colonies harboring PTZ57R-PR were confirmed by colony PCR and sequencing. Then, the blunt end DNA encoding PR sequence was PCR amplified from PTZ57R-PR and visualized on 1.5% agarose gel (Figure 1B). Sequencing result of cloned PR sequence with specific primers clarified coding sequence of CRF35_AD subtype of HIV-1 virus which is highly prevalent in Iran and Afghanistan [identities=298/299 (99%) and gaps=0/299 (0%)], except a substitution at position 262 (A>G) in our query. Since the genome of retroviruses is amplified by an error prone reverse transcriptase, the genome of these viruses is susceptible to changing during life cycle of virus. Therefore, this phenomenon is likely reasonable that why our sequencing result showed 99% homology, and not 100%. ORF analysis showed that PR coding region has been inserted in-frame so that it will be expressed next to N-terminus HP-thioredoxin fusion and C-terminus V5 epitope and (6X) His-tag.
Expression of protease recombinant protein in E. coli BL21: In TOPO-Cloning system, recombinant proteins are expressed in fusion with (6X) His-tag and HP-thioredoxin that increase the molecular weight of the product about 17 kDa. Since protease has a molecular weight of 11 kDa, it must be seen as a distinct 28-kDa band in SDS-PAGE and Western blot. Study of protein expression by SDS-PAGE and Western blot with anti-His Tag antibody showed that the protein expression of cloned rPR was performed well after induction with IPTG (Figures 2A and 2B). In SDS-PAGE analysis, a distinct band of approximately 28 kDa was seen after induction (Figure 2A), whereas there was no expression of rPR fusion protein in transformed BL21 (DE3) without IPTG induction and BL21 (DE3) without recombinant pET102D/TOPO expression vector (as negative control). SDS-PAGE analysis and subsequent Coomassie blue staining of resultant supernatant obtained from centrifugation of sonicated cell suspension indicated that recombinant protease was expressed in soluble form (Figure 2).
Purification of recombinant protease: Since the recombinant protease expression was observed in soluble form, native protein purification approach was used to maintain the structure and function of protein. Protein purification was developed using Ni-NTA affinity chromatography where the His-Tag and HP-thioredoxin are attached to nickel ion. The concentration of the purified recombinant protein measured by Micro BCA protein assay kit (PIERCE, USA) was 85 to 100 µg/ml in different dilutions. For identification and confirmation of produced rPR, Western blotting with anti-His Tag and SDS-PAGE analysis were carried out. Clear bands corresponding to 28 kDa molecular weight were observed (Figures 2A and 2B).
Specificity and sensitivity of recombinant protease in immunoblotting and ELISA assay: Western blotting and ELISA assay were performed for identification and confirmation of immunoreactiv-ity of produced rPR. Western blotting with HIV+ serum was carried out and results disclosed that produced rPR reacted specifically with antibodies which most probably were raised by the immune system against protease antigen (Figure 3).
ELISA assay was done for pre-confirmed 50 HIV+ and 50 HIV-sera. Statistical analysis was performed based on ROC analysis and Youden’s index. Our ELISA assay developed in this study could detect 45 out of 50 positive samples and was negative for 43 out of 50 negative sera, therefore, sensitivity and specificity values of rPR immunoreactivity were seen to be 90 and 86%, respectively (Table 2).
Discussion :
The protease enzyme was expressed as a fusion recombinant protein with His tag and thioredoxin in acceptable concentration of 85-100 μg/ml. In general, purification products in prior studies using E. coli system have been less than 1 mg/L and in cases with the high expression level (20-30 mg/L), reproducibility has been the problem 16-18.
In previous studies, different strategies were investigated in order to produce large amounts of HIV-1 Pr, including i) autocatalytic processing of a larger precursor ii) fusing to a variety of proteins, e.g., lactamase, Glutathione S-Transferase (GST) and maltose-binding protein fused to N-terminal portion of interferon, or as His-tagged recombinant protein iii) codon usage, A+T richness at the 5′ end of the coding region, and optimizing promoter and iv) recovering by refolding E. coli inclusion bodies 16,18-25. In these studies, producing rPR has been faced with problems such as toxicity, hydrophobicity, and purification. In this study, pET102/D.TOPO expression system was used to overcome the mentioned problems. For the production of rPR protein, the sequence from isolate IBTO-1 and PR was cloned containing region in a vector PTZ57R-PR. After sub-cloning to Pet102/D.TOPO expression vector, PR coding ORF was expressed in fusion with thioredoxin under T7 promoter control in E. coli. N-terminus thioredoxin fusion will promote protein folding, expression, and solubility as proved by others 26-30. Furthermore, increased solubility will decrease the formation of inclusion body and will ease the purification of recombinant proteins. On the other hand, the position of the cloning site in the vector used in this study is designed so that 6x histidine tag is added to the beginning and end of the protein. The existence of such histidine tags makes produced recombinant protein to be purified easily by the nickel affinity chromatography 31. Single-step purification of rPR was successfully done using nickel affinity column. The purity of the product was estimated to be more than 95% by SDS-PAGE and subsequent Coomassie blue staining (Figure 2B). In a study, Gorjipour et al also used our cloning system for production of recombinant HIV-1 P24 32. According to their results, the amount of produced protein was 120 μg/ml less than the amount of rPR production. The reason can be probably due to the highly toxic property of protease for the host.
On the other hand, diagnosis of HIV-infected individuals is a primary task that allows health providers to screen and identify the virus carriers which help prevention of infection transmission and on time treatment. Immunoreactivity of purified rPR was tested by Western blotting and ELISA. Compared with ELISA, Western blotting is more specific but less sensitive. In some studies, immunoreactivity of HIV-1 PR has showed the reactivity with sera of more than 50% of individuals infected with HIV-1 14. Bjorling et al found that HIV-1 protease has four different antigenic regions where the most prominent reactivity was resulted from amino acid 44-58 known as flap region 33. In another study, hamster monoclonal antibody showed a high specific reaction with HIV-1 protease demonstrating considerable antigenic characteristics of protease 34. Interestingly, in the study conducted by Hallengärd et al demonstrated that inactivation of protease enzyme activity significantly increases its in vitro expression as well as in vivo immunogenicity 35. Therefore, these findings are indicating that protease can be a good candidate in diagnostic and vaccine design approaches. One of the major goals of the current study was to validate the immunoreactivity of the rPR with HIV-1 infected sera. In the present study, Western blotting and ELISA assays were carried out. Our Western results revealed that produced rPR reacts with HIV-1 infected sera specifically (Figure 3). Also, ELISA data disclosed that protease is approximately a strong antigen where testing of 50 HIV-1 positive and 50 HIV-1 negative serum samples disclosed that the recombinant protein produced in this study has the diagnostic values of 90% sensitivity and 86% specificity (Table 2). So, our findings confirm the results of some previous studies.
In the present study, rPR was produced in fusion with HP-thioredoxin. In addition to enabling high level expression of recombinant fusion proteins, HP-thioredoxin has indicated the increases of coating efficiency by adsorption to the MaxiSorp MicroWell surfaces and consequently the increases in the efficiency of ELISA 32. Nonetheless, the role of fused protein tag in specificity of the final product isn’t clear and can be considered in next studies.
Conclusion :
Taking together, HIV-1 protease is one of the key enzymes that plays an important role in viral maturation so that it has been an AIDS inhibitor target for a long time 6. Nonetheless, little attention has been paid to studying the antigenic properties of protease in diagnostic and vaccine design programs. Recombinant protease in high quantity and soluble form at lab-scale was produced. Highly expressed rPR in this experiment can provide a cheap and highly qualified candidate for use in the diagnostic tests for the serodiagnosis of HIV-1 infection. Accompanied with other HIV-1 antigens, detection of antibodies using this recombinant antigen can provide a platform for screening and detection of HIV-1 infection in screening programs. Although the potency of this antigen was not studied for the induction of immune response, it is expected to be a potential target in further studies including production of monoclonal antibody and vaccine design.
Acknowledgement :
The authors thank the medical biotechnology and virology labs for helping us during the research. We also appreciate Mr. Hasannejad efforts for technical assistance. This work was supported by grant (number 90-04-31-16015) of Tehran University of Medical Sciences. The authors declare that there is no conflict of interest.
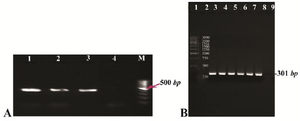
Figure 1. A) Gel agarose visualization of PCR products amplified by internal primers after amplification of cDNA as template. M (DNA marker); 1-3 (interest fragment amplified in optimized Tm=58°C); 4 (negative control, human genomic DNA); B) PCR amplification results of the PTZ57R-PR vector containing protease ORF (301 bp). Lanes 1 and 2 (DNA marker); lanes 3-6 (amplified by Taq DNA polymerase); lanes 7-8 (amplified by pfu DNA polymerase); lane 9 (negative control).
|
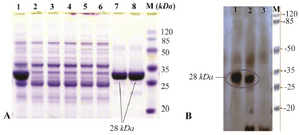
Figure 2. A) SDS-PAGE analysis of recombinant fusion thioredoxin-PR protein on 12% polyacrylamide gel. Lane M: prestained protein molecular marker (Thermo Scientific Pierce Prestained Protein MW Marker); Lane 1: sonicated BL21 (DE3) crude cell lysate harboring recombinant plasmid after induction with IPTG; lanes 7 and 8: rPR protein purified by Ni2+-NTA resin column affinity chromatography; Lanes 2-6: E. coli BL21 (DE3) lysate without recombinant vector (negative control); B) Confirmation of the recombinant PR protein production by Western blot analysis with mouse anti-HIS tag antibody. Lane 1, purified recombinant PR; lane 2, cell lysate harboring recombinant plasmid after induction; lane 3, host cell without recombinant plasmid (negative control).
|
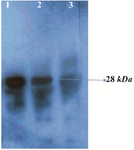
Figure 3. Western blot analysis of the rPR protein (28 kDa) with HIV-infected serum. Lane 1, purified recombinant PR; lane 2, cell lysate of E. coli harboring recombinant plasmid Pet102-PR; lane 3, host cell without recombinant plasmid Pet102-PR.
|

Table 1. Nucleotide sequence of the primers used for amplification of region containing protease gene
|
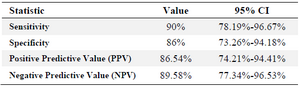
Table 2. ELISA results of 50 serum samples of the confirmed infected individuals with HIV and 50 negative serum samples as the control
|
|