Enrichment of Undifferentiated Type A Spermatogonia from Goat Testis Using Discontinuous Percoll Density Gradient and Differential Plating
-
Heidari, Banafsheh
-
Reproductive Biotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Gifani, Minoo
-
Immunology Research Center (IRC), Tabriz University of Medical Science, Tabriz, Iran
-
 Shirazi, Abolfazl
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, Tel: +98 21 22404144; Email: A.Shirazi@avicenna.ac.ir
Shirazi, Abolfazl
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, Tel: +98 21 22404144; Email: A.Shirazi@avicenna.ac.ir
-
Reproductive Biotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Zarnani, Amir-Hassan
-
Nanobiotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Baradaran, Behzad
-
Immunology Research Center (IRC), Tabriz University of Medical Science, Tabriz, Iran
-
Naderi, Mohammad Mehdi
-
Reproductive Biotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Behzadi, Bahareh
-
Reproductive Biotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Borjian-Boroujeni, Sara
-
Reproductive Biotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Sarvari, Ali
-
Reproductive Biotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Lakpour, Niknam
-
Nanobiotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Akhondi, Mohammad Mehdi
-
Reproductive Biotechnology Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
Abstract: Background: The well documented source for adult multipotent stem cells is Spermatogonial Stem Cells (SSCs). They are the foundation of spermatogenesis in the testis throughout adult life by balancing self-renewal and differentiation. The aim of this study was to assess the effect of percoll density gradient and differential plating on enrichment of undifferentiated type A spermatogonia in dissociated cellular suspension of goat testes. Additionally, we evaluated the separated fractions of the gradients in percoll and samples in differential plating at different times for cell number, viability and purification rate of goat SSCs in culture.
Methods: Testicular cells were successfully isolated from one month old goat testis using two-step enzymatic digestion and followed by two purification protocols, differential plating with different times of culture (3, 4, 5, and 6 hr) and discontinuous percoll density with different gradients (20, 28, 30, and 32%). The difference of percentage of undifferentiated SSCs (PGP9.5 positive) in each method was compared using ANOVA and comparison between the highest percentage of corresponding value between two methods was carried out by t-test using Sigma Stat (ver. 3.5).
Results: The highest PGP9.5 (94.6±0.4) and the lowest c-Kit positive (25.1±0.7) in Percoll method was significantly (p≤0.001) achieved in 32% percoll gradient. While the corresponding rates in differential plating method for the highest PGP9.5 positive cells (81.3±1.1) and lowest c-Kit (17.1±1.4) was achieved after
5 hr culturing (p<0.001). The enrichment of undifferentiated type A spermatogonia using Percoll was more efficient than differential plating method (p<0.001).
Conclusion: Percoll density gradient and differential plating were efficient and fast methods for enrichment of type A spermatogonial stem cells from goat testes.
Introduction :
Type A Spermatogonial Stem Cells (SSCs) are small self-renewing subpopulation of undifferentiated spermatogonia that are defined by their unique properties such as prolonged proliferation, self-renewal, generation of differentiated progeny, and maintenance of developmental potential 1. These cells can proliferate rapidly when the testis is damaged by chemicals or radiation, whereas under physiological conditions, these cells divide slowly to produce both stem cells and progenitor cells 2,3. When transplanted into the seminiferous tubules of an infertile male, they can establish donor-derived spermatogenesis and produce spermatozoa that transmit the donor haplotype to progeny 4. Additionally, when they are cultured in appropriate conditions, they can acquire pluripotency and differentiate into derivatives of the three embryonic germ layers including sperm 5,6.
A great excitement and expectation in today’s biomedical world is the study of stem cells, owing to their ability to exist in an undifferentiated state and their transformation into different tissue types, depending on the ambient conditions 7.
The complexity of the seminiferous epithelium and systemic and testicular microenvironments makes it difficult to study cellular and molecular aspects of the regulation of spermatogonial stem cell behavior 8,9. This deficiency is largely caused by the small number of SSCs in testis and the inability to isolate a purified SSCs population because of the lack of specific, accurate and inexpensive purification protocol with minimum side effects on spermatogonia 9.
It is valuable to develop an efficient method for SSCs enrichment as well as in vitro culture of purified SSCs and modulate culture conditions that control self-renewal versus differentiation in vitro. The practice would allow us to better understand the various aspects of adult stem cell biology such as the mechanisms that regulate SSC renewal and differentiation, the cell signaling pathways that regulate SSC function, to unravel the cellular and molecular mechanisms driving spermatogenesis, to preserve and manipulate male fertility and tissue regeneration, and to develop more improved techniques for male germline modification and transfection 10-13.
Additionally, due to extremely small population of SSCs as the stem cells that transmit genetic information to subsequent generations (for instance type A spermatogonia comprise 0.02-0.03% of total germ cells) 14-16, the ability to study their self-renewal and their characteristics is limited and further investigation requires larger populations of pure SSCs 17. Several laboratories have established the methods which allow the enrichment of male germline stem cells or improve their culture conditions such as expansion techniques with feeder layers or a combination of growth factors, or serial transplantation procedures 10,18-20.
In 1970s, the feature of serpmatogonia was known better 20,21. Bellve et al (1977) first used BSA to isolate spermatogonia from 8 day old mice and obtained a 90% purified fraction of type A spermatogonia 22. Bucci et al (1986) used enzymatic digestion and got 76% purity of spermatogoina from rat testis 23. But these methods either used a large number of experimental animals or resulted in insufficient purity of spermatogonia for experimental analyses.
With improvements in cellular biology approaches during several decades, the technology of isolation and purification of spermatogonia achieved a great breakthrough. Several methods have been employed to isolate SSC in different species including elutriation 23, differential plating 7,17,24, velocity sedimentation 10,22, discontinuous percoll density gradient 25,26, Magnetic-Activated Cells Sorting (MACS) 7,10,13 and Fluorescence Activated Cells Sorting (FACS) 26,27.
Considering the inadequate information regarding the effect of purification protocols on goat type A SSCs and the application of transfected SSCs as an efficient tool in production of transgenic animals, the present study was designed to compare the differential plating and percoll density gradient methods in enrichment of undifferentiated spermatogonia in goats.
Undoubtedly, availability of best practical isolation and purification techniques and establishment of stable SSCs in vitro will pave the way for innovative research into stem cell biology, leading to further breakthrough in the understanding of spermatogonial physiology and development of powerful tools for fertility preservation.
Materials and Methods :
Except where otherwise indicated, all chemicals were obtained from Sigma (St. Louis, MO, USA).
Collection and evaluation of testis: Following castration of a one month old goat, the testes were transported to the lab in transition media (PBS+antibiotics) at 37C. After macroscopic evaluation of the testes for any pathologic signs (trauma, cyst, tumor and hematoma), the testes were washed (3 times) with transition media and the tunica albuginea and visible connective tissues were aseptically removed.
Cell isolation and preparation: Single cell suspensions were prepared using a protocol which was recently described 28. Briefly, upon arrival of testes to laboratory, they were washed several times with transition media and tunica albuginea and visible connective tissues were removed. Testes samples were transferred into the Dulbeco Modified Essential Medium (DMEM) supplemented with NaHCO3 (14 mol/l), Hepes (15 mol/l), NEAA (10 µl/ml), penicillin (50 IU/ml) and streptomycin (50 mg/ml) for 5-8 min. Then testis was again washed in sterile transition media and dissection was done aseptically under laminar airflow hood. The SSCs were isolated through two-step digestion methods. The samples were incubated in DMEM containing 1 mg/ml collagenase type I at 38C in 5% CO2 for 60 min. The samples were examined with an interval of 1 min for determination about disentanglement. After initiation of tissue disentanglement, the somniferous tubules’ fragments were separated from interstitial cells by centrifugation at 100×g for 1 min, and the supernatant, containing mostly the interstitial cells, was discarded. The tubules’ fragments were washed two times with ice-cold DMEM.
In a second digestion step, the tubules’ fragments were incubated in trypsin/EDTA (0.25% /1 mM) for 12-25 min. The dispersed cells were then washed twice in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS; GibcoBRL) to stop the enzymatic digestion and undigested debris was removed by centrifugation at 100×g for 1 min. The supernatant was then processed by sequential filtration through 60 µm nylon mesh (Small part, F062N-08-C). The filtrate was centrifuged at 500×g for 5 min, and the pellet was resuspended in DMEM supplemented with 10% FBS (Gibco) and NEAA. In the final cell suspension, the different testicular cell types including Sertoli cells and spermatogonia with different sizes were identified using light microscope. Total cell number and cell viability were determined after trypan blue staining. The cell suspension was mixed with 0.4% trypan blue (1:1, v/v). The number of live (trypan blue excluding) and dead cells were determined using hemocytometer.
Identification of SSC by histological evaluation: For histological evaluation, a small sample of testes was fixed overnight in Bouin’s solution, washed in 70% ethanol, embedded in paraffin and sectioned at 5 μm using standard procedures. The sections were stained with Hematoxylin and Eosin and examined under a light microscope for identification of cell types and their developmental stages.
In cell suspension, after collection of SSCs at each stage, SSCs were identified based on their morphology in phase contrast microscopy.
Enrichment of goat undifferentiated SSCs by discontinuous percoll density gradient: The spermatogonial stem cells were enriched from cell suspension by following the method of Gang et al 29 with minor modification. Briefly, percoll was first sterilized by autoclaving and diluted to gradient concentrations of 20, 28, 30 and 32% with PBS. The gradient was made in 15 ml graduated tube by adding 1 ml of each of percoll solution with different densities in a sequence that the highest density percoll solution came in bottom and that of the lowest in the top of the tube. The newly collected cell suspension was slowly layered on the top of the above gradient and centrifuged at 800×g for 30 min at 18ºC. The cells were collected in the interface between different densities of percoll solution, washed twice with PBS and resuspended in culture medium (1 ml). The cell numbers and cells viability were determined by trypan blue staining. The cell suspension was mixed with 0.4% trypan blue (1:1, v/v). The number of live (trypan blue excluding) and dead cells were determined using hemocytometer. The collected cells, in each gradient, were cytospun at 400 rpm for 5 min in poly-L-lysine pretreated slides (5×104 cells per each slide). Two immunocytochemical reactions were successively performed using PGP9.5 and c-kit primary antibodies.
Enrichment of undifferentiated SSCs by differential plating: The freshly isolated cells were seeded at a concentration of 2×104/cm2 in 12-well cell chamber slides (Falcon, USA) in basic culture system consisted of high glucose DMEM (GibcoBRL) supplemented with 10% FBS and 1% penicillin-streptomycin (GibcoBRL). The cells were incubated in atmosphere of 5% CO2 with 85% relative humidity at 37°C for 3, 4, 5 and 6 hr. After that, the culture solutions containing non-adherent cells were removed gently and centrifuged at 1200 rpm for 10 min. The cell numbers and viability rates were determined after trypan blue staining. The cell suspension was mixed with 0.4% trypan blue (1:1, v/v). The collected cells, in each time, were cytospun at 400 rpm for 5 min in poly-L-lysine pretreated slides (5×104 cells per each). Two immunocytochemical reactions were successively performed using PGP9.5 and c-kit.
Identification of SSCs and purity detection by immunocytochemical staining: Type A spermatogonia were identified through immunocytochemical staining according to the protocol described by Herdari et al 28. Briefly, the purified cells, 5×104 cells, in each group were placed in each slide and centrifuged at 400 rpm for 5 min, fixed in acetone for 2 min at -20°C and placed at 4°C for 2 min. After drying the slides, cells were immunostained as described. Antigen retrieval was performed on sections through immunostaining for PGP 9.5 and c-kit by Tween 20 (0.2% in PBS). The first unspecific site blocking was done with avidin/biotin. Therefore, the slides were incubated with avidin for 10 min in dark, and after three times of washing were incubated with biotin for 10 min in the same condition. All sections were exposed to 0.3% H2O2 for 15 min in dark to inhibit endogenous peroxidase and washed in TBS/BSA (three times, 4 min each).
Another unspecific site blocking was done with 10% sheep serum in PBS for 30 min at room temperature. Subsequently, the slides were incubated with unconjugated primary antibodies including rabbit anti-PGP 9.5 (Dako, Carpinteria, CA, USA) and rabbit anti c-kit (Santa Cruz, Santa Cruz, CA, USA), each used at 1:100 dilution in PBS with 2.5% goat serum (PBS-GS), for 1 hr at room temperature. After washing three times in TBS/BSA (5 min each), the sections were exposed to secondary antibody (biotinylated sheep anti-rabbit IgG, Avicenna Research Institute, Iran) for 45 min at room temperature and washed in TBS/BSA as described above. The sections were exposed to HRP-conjugated streptavidin (Biosource, USA) with 1:500 dilution for 30 min, and then washed with TBS/BSA. At the final step, color was developed by the addition of 3, 3’- Diaminobenzidine (DAB; Roche, Germany) for 8-10 min. The slides were rinsed thoroughly in distilled water, counterstained with Harris hematoxylin for 30 s, washed in distilled water, dehydrated in graded alcohols, cleared in xylol, and were then mounted in Entellan (Merck, Germany).
Immunohistochemistry results were quantified by two evaluators blinded to the diagnosis of immunohistochemical reactivity. The percentages of PGP9.5 and c-kit positive and negative cells were evaluated by cell counting of prepared immunocytochemical slides.
Statistical analysis: All data were analyzed using one way ANOVA (Tukey Test). A p<0.05 was defined as statistical significance.
Results :
Histological evaluation of testes: In histological evaluation, the immature Sertoli cells, spermatogonial cells, and a small number of leydig cells were identified by morphological evaluations (Figures 1A and B). The seminiferous tubules had no lumen and different subpopulations of type A spermatogonia were mostly localized in the middle of the somniferous tubules (Figures 1A and B).
In seminiferous tubules, the presence of three groups of spermatogonia with different sizes was determined (Figures 1A and B). These cells were often round or oval in shape with the following characteristics: normal size of about 10-12 m in diameter, big spherical nucleus containing one to three irregular nucleoli of about 1-3 µm in diameter, high nucleus, cytoplasm ratio, and cytoplasmic inclusions which were mostly concentrated at one side of the cell (Figures 1 and 2).
Cytological analysis of spermatogonial stem cells: After cellular isolation, the phase contrast microscopy was used to identify spermatogonial stem cells in fractions on the basis of SSCs morphology. The cellular suspension was consisted of relatively homogenous population of the cells.
The presence of four groups of undifferentiated type A spermatogonia with different sizes in cell suspension was confirmed (Figure 2). They were observed in different forms as single (Figure 2A), paired (Figures 2B and C), aligned and cluster (Figure 2D) forms. The isolated SSCs, with more than 95% viability tended to congregate and form small cell clusters.
Enrichment of type A spermatogonia by discontinuous percoll gradient: The percentage of type A spermatogonia in suspension was taken to determine the effectiveness of the gradients on enrichment of the undifferentiated spermatogonia.
Data indicated the significant differences in the percentage of type A spermatogonia in cell population across the individual gradients (p<0.001) and the percentage of PGP9.5 positive cells (undifferentiated type A cells) was found to be significantly different among groups. The proportions of undifferentiated spermatogonia in 30 and 32% gradients were significantly higher than 20 and 28% gradients (p<0.001). The lowest percentage of undifferentiated type A spermatogonia was determined in 20% gradient (Figure 3). Concurrent with the increment of percoll gradient, the number of differentiated type A spermatogonia (c-kit positive cells) decreased (p<0.001, Figure 4). The highest and lowest percentages of undifferentiated spermatogonia were found in gradients 32 and 20%, respectively (Table 1). Cell viability in all fractions was observed and found to be almost identical (100 %).
Enrichment of type A spermatogonia by differential plating: According to the expression of PGP9.5 and c-kit molecular markers, the optimum time for adherence of most of somatic cells to plate and having the highest percentage of undifferentiated type A spermatogonia was 5 hr after culture initiation (Table 1).
Enrichment of cell suspension, by 5 hr of culturing, resulted in a significant increase in PGP9.5 positive cells and reduced the population of differentiated type A spermatogonia (p<0.001, Figures 5 and 6).
Discussion :
Spermatogonial stem cells are unipotent adult stem cells responsible for maintenance of the fertility throughout the entire life of the males. These cells are the foundation of spermatogenesis 11 and have the potential to self-renew and at the same time generate the cascade of differentiating germ cells that will eventually lead to the formation of sperm 12,17.
The development of the methods for the isolation and purification of undifferentiated SSCs in vitro will be the key in identification of their biochemical and morphologic properties, and attainment of the great potentials of SSCs in development of novel therapeutic strategies for treatment of infertility 1,13,30. The important aspects of spermatogonial stem cells resulted in their widespread use in reproductive technologies such as spermatogonial stem cell transplantation and transgenesis in species wherein embryonic stem cells are not available and somatic cell nuclear transfer and reprogramming pose several problems 16,30,31.
Three types of spermatogonia (types A, intermediate, and B) were initially described based on their nuclear morphology 32. Spermatogonial stem cells undergo both self-replicating proliferation and production of daughter progeny to become type A spermatogonia 16,17. The main characteristics in analysis of goat type A spermatogonia were morphological characteristics of the cells (nuclear and nucleolus) and the expression level of specific markers 28. In previous study, the presence of three groups of type A spermatogonia with different morphological characteristics in seminiferous tubules by immunostaining techniques was determined. Basal stem cells (As, Ap spermatogonia) were observed as the small round cells with thin rim of cytoplasm, large central nucleolus and one to three irregular nucleoli. Aggregated spermatogonia (Aal) with different sizes from small to large, containing one to two nucleoli and the larger ones committed spermatogonia that consisted of subpopulations of differentiating spermatogonia (A1-A4) 28. The difference in size of the spermatogonia might be related to either the phase of cell cycle 33 or their differentiation status 28,34.
The testicular suspension isolated from seminiferous tubules is often contaminated by differentiating spermatogonial cells, Sertoli cells, Leydig cells and peritubular myoid cells 11,13. Among the large population of somatic cells and differentiating germ cells within seminiferous tubules, only a small population of undifferentiated type A spermatogonia resides at the basement membrane of seminiferous tubules 7. Most culture systems are known to contain a mixture of testicular cells with about 1.33% SSCs 11,12,17. Therefore, the establishment of an efficient purification technique with the least influence on the SSCs survival and proliferation is necessary.
Among different methods of goat type A spermatogonia purification, in the present study, percoll density gradient and differential plating were chosen because these methods were faster, easier, less costly, safer and had the least negative effects on SSCs viability and their morphological characteristics.
The low viscosity and osmolality, two ideal features for separation of various cell types, has made the percoll ideal for separation of various types of cells 35. Percoll has been applied to separate adult and prepuberal types of spermatogonia cells in chicken 36, porcine 37, bovine 34,35, and murine 25. The cell viability using percoll in previous studies 25,38 ranged from 30 to 95% after SSCs separation which was consistent with our results (100%).
In the present study, the maximum type A spermatogonia purification (94.6%±0.4) was achieved by 32% percoll density gradient that was slightly higher than those studies which applied percoll in other species including rat with 80±6.1% SSCs purification in 30%-32% gradient 25, pig with 83.62±4.24% SSCs purification in 30-45% gradient 37, and human with 86.7% SSCs purification in 27-35% gradient 9.
In SSCs purification using differential plating, during cellular incubation, the somatic cells were attached to the bottom of culture plate due to its anchorage dependence so that the fast adherent velocity of the Sertoli and Leydig cells makes it possible for the majority of them to be removed from cell suspension after differential plating. In the present study, the maximum purity of undifferentiated type A spermatogoinia (81.3%) in differential plating was achieved 5 hr after culture initiation which was identical to the report by Kokkinaki with a purity of 85.7% obtained for mouse testicular tissue 38. The obtained purity, however, in rat (97%) and pig (94%) using this method with 90-95% viability rate 30,39 was higher than the corresponding rate in the current study. Concerning the time of culturing, the overnight incubation in other species resulted in lower purification rate compared to our results, for instance, 40% in bovine 17 and 75% in ram 40. In above studies, the cellular viability after isolation was also lower than the one in the present study (80-90% vs. 99%).
Conclusion :
In our study, apart from more typical morphological characteristics of SSCs and the higher intensity of immunocytochemical reactivity in percoll method compared to differential plating, regarding the superiority of two purification methods in separation of undifferentiated spermatogonia, higher PGP9.5 positive and lower c-kit positive, the maximum purification rate in percoll method (94.6%) was significantly (p<0.001) higher than the maximum rate in differential plating method (81.3%). The higher purity of undifferentiated spermatogonia using percoll compared to differential plating method in ovine and bovine species 35,40 was in agreement to the present results.
Acknowledgement :
The authors would like to thank Avicenna Research Institute for technical and financial
supports, ACECR, Tehran.
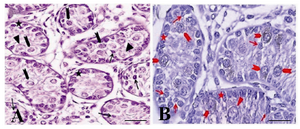
Figure 1. The immunohistochemical evaluation of the testis of a one month old goat using an antibody against c-kit. The presence of three groups of spermatogonia with different sizes was determined: basal (arrow, A, B) aggregated (arrowhead, A, B) and committed (triangle, A, B) type A spermatogonia. Both aggregated and committed sperm-atogonia were c-kit positive. Note that the strong and weak c-kit immunoreactivity was observed among aggregated (arrowhead, A, B) and committed (triangle, A, B) respectively. Basal spermatogonia (arrow, A, B), surrounding Sertoli and Leydig cells were negative for c-kit
|
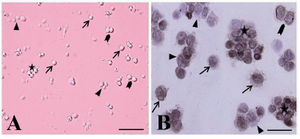
Figure 2. The cytological and immunocytochemical evaluation of spermatogonial stem cells using PGP9.5 immediately after isolation. The spermatogonia were seen as different forms: single (arrow, A, B), paired (arrowhead, A, B) align-ed (triangle, A, B) and cluster (asterisk, A, B). The isolated SSCs were round cells with a spherical nucleus, a high nucleus, cytoplasm ratio, and many cytoplasmic inclusions that tended to congregate and form small cell clusters
|
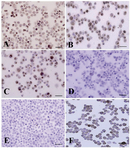
Figure 3. Immunocytochemical identification of undifferentiated type A spermatogonia after percoll separation method using an antibody against PGP9.5. Basal spermatogonia (dark brown) and some aggregated spermatogonia (light brown) in gradients A) 32% and B) 30% were significantly higher than C) 28% and D) 20% gradients. Note the intense staining in the cytoplasm, especially in the vicinity of the nucleus, in basal spermatogonia. E) Negative and F) positive controls were detected
|
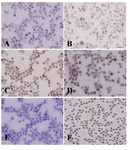
Figure 4. Immunocytochemical evaluation of differentiated type A spermatogonia in percoll separation method using c-kit antibody. The proportion of c-kit positive cells (brown cells) in A) 32%; B) 30%; C) 28% and D) 20% gradients were determined. The highest (D) and lowest (A) percentages of differentiated spermatogonia were found in gradients 32 and 20% respectively. Note that the strong and weak c-kit immunoreactivity was observed among the aggregated (Aal) and committed (A1-A4) spermatogonia, respectively. E) Negative and F) positive controls were detected
|
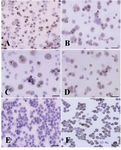
Figure 5. Immunocytochemical localization of differentiated type A spermatogonia in differential plating method using
c-kit. The percentage of c-kit positive cells (Aggregated sperm-atogonia, dark brown and committed spermatogonia, light brown) was evaluated; A) 3 hr; B) 4 hr; C) 5 hr and D) 6 hr after culture initiation. The most purified undifferentiated spermatogonia were achieved 5 hr after culture initiation. E) Negative and F) positive controls were detected
|

Figure 6. Immunocytochemical identification of undifferentiated type A spermatogonia by differential plating method using PGP9.5. The proportion of PGP9.5 positive cells (basal spermatogonia, dark brown, and some aggregated sperm-atogonia, light brown) C) 5 hr after culture initiation was significantly higher than; A) 3 hr; B) 4 hr and D) 6 hr after culture. E) Negative and F) positive controls were determined
|
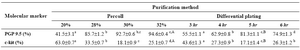
Table 1. Two methods of goat SSCs purification using PGP9.5 and c-kit molecular markers
a-d: In each method numbers with different lowercase letters in the same row differ significantly (p<0.001)
aA, bB: Numbers with different uppercase letters in the same row column indicates significant difference between two methods (p<0.001)
|
|